The radiochemical synthesis of [18F]3F4AP comprises two steps (Figure 1). The first step is carried out in a fully automated fashion using the synthesis unit (Figure 3). This cassette-based system uses four reagent vials and one reactor vial and has computer-controlled valves that allow transfer and mixing of reagents as well as heating, pressurizing and evacuating the reactor. In addition, it supports standard solid-phase extraction cartridges for separation of reagents. The computer interface allows users to write and modify scripts in order to run their own syntheses. In the case of [18F]3F4AP, the synthesis procedure is comprised of five basic parts. In the first part, the synthesizer performs self-check steps, preheats the reactor and waits for operator's signal that the 18F is ready. During the second part, the [18F]fluoride is transferred from the 18F vial into the anion exchange cartridge and eluted from the cartridge into the reactor using a solution tetrabutyl ammonium bicarbonate. The third part, the synthesizer azeotropically dries the [18F]fluoride under vacuum to make it reactive towards nucleophilic displacement. In the fourth part, the precursor is automatically added to the reactor where it reacts with the 18F– to generate the labeled compound. Finally, the reaction is quenched by the addition of 0.2% oxalic acid in methanol, which prevents base-promoted decomposition of the product, and the final solution is pressure-transferred to the collection vial after passing through an alumina-N cartridge that traps any unreacted fluoride.
After the labeling step is completed a small sample can be taken for quality control. Running a sample on the HPLC provides confirmation that the labeling step worked and an estimation of the radiochemical purity (Figure 4). Also, from the UV trace on the HPLC the mass amount of product can be calculated using a pre-established calibration curve.
While the in-process quality control HPLC is running, the second reaction step, reduction of the N-oxide and nitro groups, is performed. In order to do this, the labeled product is automatedly injected into an in-house hydrogenation device based on the method published by Yoswathananont et al.13 (Figure 2). This device consists of an HPLC pump and a compressed hydrogen tank connected to the flow hydrogenation device through lines equipped with check valves to prevent back-streaming. The product is pushed by the HPLC pump and mixed with hydrogen in a T-shaped mixer. This mixture is then passed through a small cartridge containing 10% Pd/C catalyst on a solid support. After passing through the catalyst the reduced product is then collected in small fractions.
Following hydrogenation, the crude product is transported and manually injected into the HPLC for purification of the final product (Figure 5). The mobile phase of the HPLC has been selected to be compatible with animal injection. The peaks corresponding to the product are then collected and filtered-sterilized to obtain the final dose.
Prior to releasing the dose for PET imaging studies, quality control tests are performed. These tests are performed to ensure that the tracer is the chemical entity that it is supposed to be and that it is safe for injection. Some of these tests may not be required for injection into animals but it is generally recommended to follow the human use guidelines. Doing so ensures quality of the product, which increases confidence in the results and greatly facilitates future transition to manufacturing the product for human injection.
Table 1 contains the typical synthesis parameters including initial amount radioactivity, initial amount of precursor, yield for each step, specific activity, filtering loses, etc. These parameters are useful troubleshooting occasional failures and future optimization of the procedure.
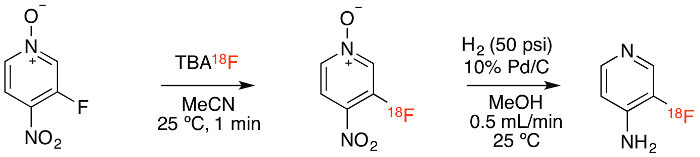
Figure 1. Reaction scheme. Radiochemical synthesis consists of labeling by 19F/18F exchange followed by palladium-catalyzed hydrogenation. Please click here to view a larger version of this figure.
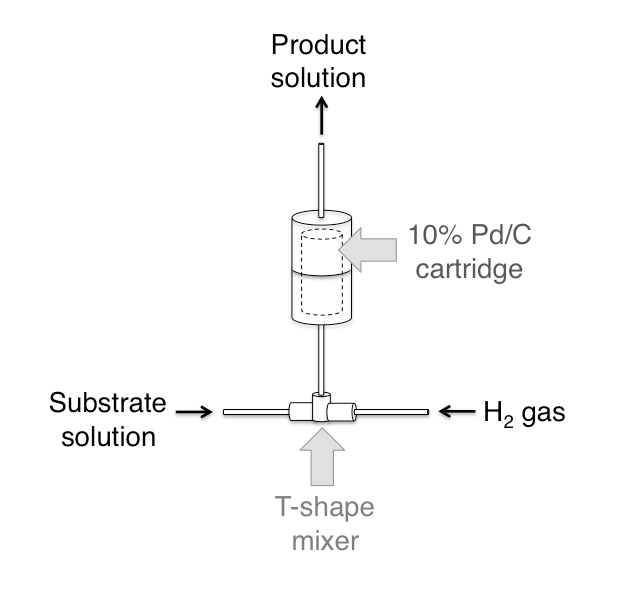
Figure 2. Hydrogenation system. Schematic of the device. This device is based on the publication by Yoswathananont et al. (ref 13).
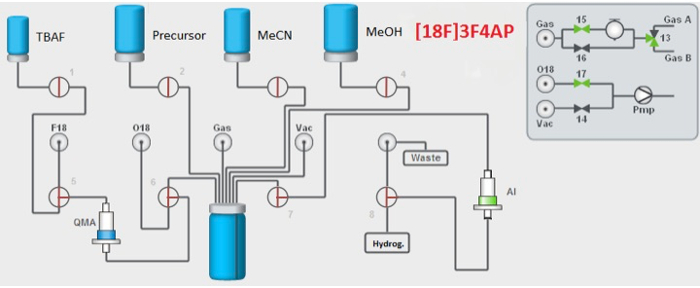
Figure 3. Scheme of synthesizer integrated fluidic processor (IFP) and reagents. IFP contains four reagent vials, a QMA cartridge and one reactor vial. Please click here to view a larger version of this figure.
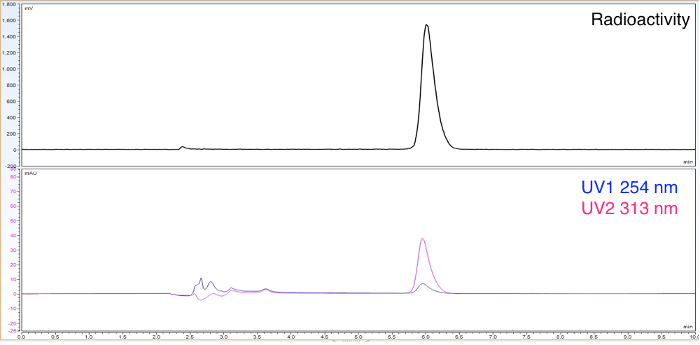
Figure 4. UV and radioHPLC tracers for intermediate product. 3-fluoro-4-nitropyridine N-oxide has a characteristic absorption at 313 nm. Please click here to view a larger version of this figure.
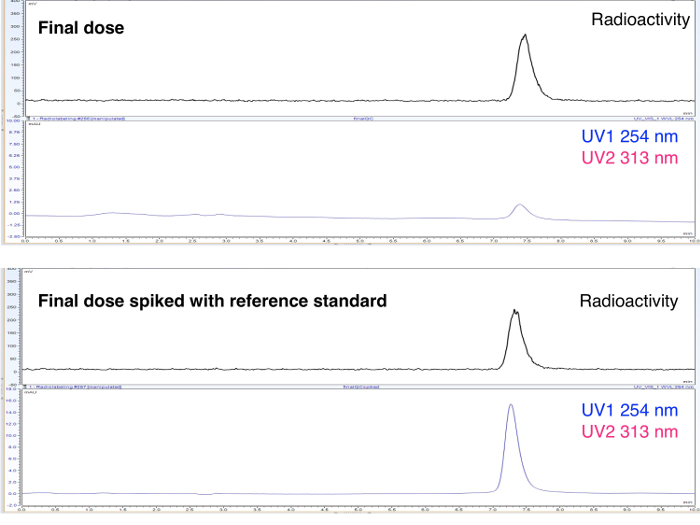
Figure 5. UV and radioHPLC tracers for final product. 3-fluoro-4-aminoopyridine absorbs at a 254 nm. Please click here to view a larger version of this figure.
| Concept | Mean (n = 4) | S.D. | Comments |
| Initial 18F activity (mCi) | 148.0 | 44.9 | Start of synthesis |
| Precursor amount (μg) | 50 | Use 50 μL of 1.0 mg/mL stock | |
| Activity left in QMA (mCi) | 3.0 | 1.7 | Measured at the end of labeling step |
| Radiolabeling yield | 29.7% | 6.3% | Act_collection_vial ÷ (Act_collection_vial + Act_AluN) |
| Radiochemical purity (HPLC-1) | > 98% | From HPLC-1 QC | |
| Spec. act. intermediate (mCi/μmol) | 122.9 | 29.7 | From HPLC-1 using calibration curve |
| Hydrogenation recovery (d.c.) | 74% | 9.0% | Corrected for decay |
| HPLC radiochemical purity (HPLC-2) | 90.7% | 2.9% | Calculated from HPLC-2 |
| Drying efficiency | > 98% | Corrected for decay | |
| Filtering recovery | 93.5% | 1.7% | Corrected for decay |
| Dose volume (mL) | 3.3 | Collect fractions with highest radioactivity | |
| Spec. act. final product (mCi/µmol) | 75.5 | 30.0 | From HPLC-3 using calibration curve |
| Synthesis efficiency | 8.5% | 3.6% | Non-decay corrected |
| Synthesis time (min) | 104 | 11.2 |
Table 1. Radiochemical synthesis parameters.
| Common problems | Potential reasons and solutions |
| [18F]fluoride is not efficiently eluted from the QMA | · TBA-HCO3 was not prepared correctly. Ensure the concentration is adequate. |
| · There are leaks on the TBA-HCO3 vial. Make sure the crimp seal is tight and the septum is not pierced prior to installing it on the IFP. | |
| · TBA-HCO3 is not in good condition. Order a fresh batch. | |
| Labeling yield is low | · There is moisture in the precursor solution. Dry precursor and solvents. |
| · Temperature is too low. | |
| Reaction solution is yellow | · The product is decomposing due to base. Use less TBA-HCO3. |
| · There is too much precursor. Use less precursor. | |
| · There is too little solvent for the amount of 18F–. Use more solvent. | |
| Additional peaks on radioHPLC | · Nitro group is being substituted: reduce the reaction temperature or shorten reaction time. |
| Hydrogenation reaction does not work | · Catalyst is not good. Use a new cartridge. |
| · Flow is too fast and does not allow sufficient contact between catalyst and substrate. Decrease flow. | |
| · Hydrogen pressure is too low. Increase H2 pressure. | |
| Hydrogen pressure increases dramatically during procedure | · Cartridge integrity is compromised and solid support is clogging the lines. Stop the flow and shut off the gas. Let radioactivity decay. Remove catalyst cartridge and flush the system. Put a new cartridge. |
| Hydrogenation yield is low | · Too many impurities competing for the catalyst (MeCN, oxalic acid). Decrease amount of impurities or increase mass of precursor (Warning: increasing precursor amount will reduce specific activity). |
| Recovery of radioactivity from hydrogenation step is low | · There is a leak in the system. Check for leaks and backflush into the hydrogen line. |
| · Compound is defluorinating in the reactor. Evaluate different reaction conditions (pressure, temperature, flow, etc.). | |
| Too much radioactivity is lost during filtration | · Wet the filter prior to use. |
| · Use filter with a lower dead volume. | |
| The final product peak on the HPLC looks broad | · Too much volume injected. Inject lower amount. Use column with larger diameter. |
| · The column is not well conditioned. Condition the column for at least 30 column volumes. | |
| · pH of the mobile phase is low. Make sure that the pH ≥ 8. | |
| · Column is not in good condition. Replace column. Use column compatible with basic pH. |
Table 2. Troubleshooting guide.
