Adenylate kinase (Adk) is a ubiquitous enzyme that functions to maintain the equilibrium between cytoplasmic nucleotides essential for many cellular processes. Adk operates by catalyzing the reversible transfer of a phosphoryl group from ATP to AMP. This reaction is accompanied by well-studied rate limiting conformational transitions 3,21. Here we analyze all currently available Adk structures with Bio3D-web to reveal detailed features and mechanistic principles of these essential transitions.
We can begin our Bio3D-web analysis of Adk by entering the RCSB PDB code of any known Adk structure. For example, entering the PDB ID 1AKE in panel A of the SEARCH tab returns 167 sequence similar structures from which the top 26 are automatically selected for further analysis (see panel B). The annotation presented in panel C indicates that these selected structures are all from E. coli, were solved by x-ray diffraction in a range of space groups; have a resolution range of 1.63 to 2.8 Å, and were co-crystalized with a range of different ligands (including no ligands, AMP, ADP, MG and the inhibitor AP5). Note that additional annotation details can be displayed by clicking on "Show/Hide Columns" option in panel C.
Multiple sequence alignment is performed upon entering the ALIGN tab. The first panel of the ALIGN tab displays a summary of the alignment providing details on the number of sequence rows (equivalent to the number of PDB structures), as well as the number of positions (i.e. alignment columns). This includes a specification of the number of gap and non-gap containing columns. The figure on the right hand side of the first row provides a schematic representation of the sequence alignment. Here the grey areas represent non-gap positions, while white areas in the alignment correspond to gaps. A representation of the sequence conservation is shown above the alignment with red areas indicating well-conserved positions, and white indicating less conserved. Note that the sequences in this figure are ordered based on their similarity provided by the clustering dendrogram on the left hand side. The second panel of this tab further facilitates clustering of the selected PDBs based on their pair-wise sequence similarity, which can be visualized either as a dendrogram or a heat map. By default, a dendrogram (or tree diagram) representing the arrangement of clusters is shown. The y- axis of the dendrogram represents the distance (in terms of sequence identity) between the clusters.
Structure superposition is performed automatically upon entering the FIT tab. The superimposed structures, displayed interactively in panel A, indicate the presence of a relatively rigid core region (encompassing residues 1-29, 68-117, and 161-214; see the 'optional core and RMSD details' panel at the bottom of the FIT tab for details). Two more variable nucleotide-binding regions (residues 30-67 and 118-167) are also clearly visible (Figure 2). RMSD-based clustering groups these structures into two distinct conformations.
Clicking on the PCA tab more clearly shows the relationship between the structures in terms of the displacements of these regions that effectively close over the bound nucleotide species in related structures (Figure 2B and 2C). The majority of structures are in the 'closed' form (blue in Figure 2C) and are associated with a bound ligand or inhibitor. In contrast more 'open' conformations are nucleotide and inhibitor free. This is consistent with the extensive body of research on Adk structure and dynamics indicating that an open configuration of these regions is required for nucleotide binding and a closed conformation for efficient phosphoryl transfer and suppression of detrimental hydrolysis events. It is notable that a single PC captures 97% of the total mean square displacement in this Adk structure set and provides a clear and compelling description of the open to closed transition along with the individual residue contributions to this functional displacement (panel C of the app and Figure 2).
Visiting the NMA tab and increasing the number of structures considered for calculation (via decreasing the cutoff for filtering similar structures) indicates that open state structures display enhanced local and global dynamics in comparison to the closed form structures (Figure 2D and panel C of app). Comparing PCA and NMA results for individual structures (panel D) indicates that the first mode of all open form structures displays a relatively high overlap to PC1 (with a mean value of 0.37 ± 0.04). In contrast, closed form structures display lower values (with a mean of 0.30 ± 0.01). RMSIP values for open form structures (0.62 ± 0.003) are also higher than those of closed structures (0.56 ± 0.008). In addition, overlap analysis shows that the first modes of the open state are in good agreement with the conformational change that describes the difference of the open and closed states (panel E). Clustering based on RMSIP values again displays a consistent partitioning of open and closed state structures (panel F).
Collectively these results indicate the existence of two major distinct conformational states for Adk. These differ by a collective low frequency displacement of two nucleotide-binding site regions that display distinct flexibilities upon nucleotide binding.
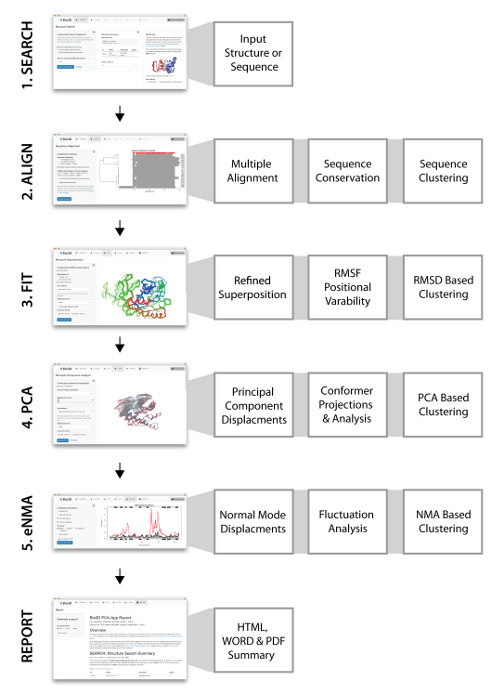
Figure 1: Bio3D-web overview with screen shots of the PCA and NMA tabs. Bio3D-web takes a user provided protein structure or sequence as input in the SEARCH tab (1). The server provides a list of related structures, which can be selected for further analysis. (2) The ALIGN tab provides sequence alignment and analysis of the structures selected in the SEARCH tab. (3) In the FIT tab all structures are superimposed and visualized in 3D together with the results of conventional pair-wise structure analysis. (4) Principal component analysis of the structure set is performed in the PCA tab to characterize inter-conformer relationships. (5) Normal mode analysis on each structure can be carried out in the eNMA tab to explore dynamic trends for the available structural states. Please click here to view a larger version of this figure.
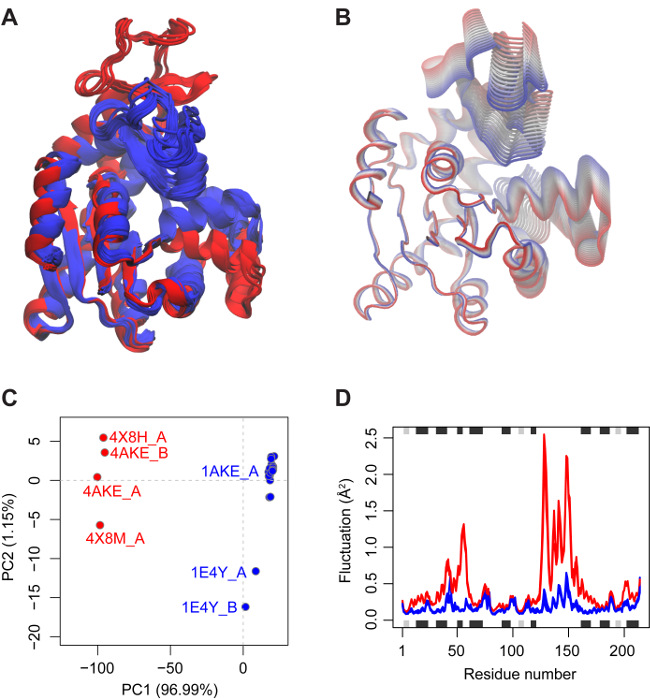
Figure 2: Results of Bio3D-web analysis of adenylate kinase. (A) Available PDB structures of adenylate kinase superimposed on the identified invariant core. Structures are colored according to RMSD-based clustering provided in the FIT tab. (B) Visualization of the principal components is available from the PCA tab to characterize the major conformational variations in the data set. Here, the trajectory corresponding to the first principal component is shown in tube representation showing the large-scale closing motion of the protein. (C) Structures are projected onto their two first principal components in a conformer plot showing a low-dimensional representation of the conformational variability. Each dot (or structure) is colored according to user specified criteria, in this case PCA-based clustering results. (D) Normal mode analysis in the eNMA tab suggests enhanced local and global dynamics for structures in the open state (red) in comparison to the closed form (blue) structures. Please click here to view a larger version of this figure.
