G2-seq: A High Throughput Sequencing-based Technique for Identifying Late Replicating Regions of the Genome
Summary
We describe a technique for combining flow cytometry and high throughput sequencing to identify late replicating regions of the genome.
Abstract
Numerous techniques have been developed to follow the progress of DNA replication through the S phase of the cell cycle. Most of these techniques have been directed toward elucidation of the location and timing of initiation of genome duplication rather than its completion. However, it is critical that we understand regions of the genome that are last to complete replication, because these regions suffer elevated levels of chromosomal breakage and mutation, and they have been associated with both disease and aging. Here we describe how we have extended a technique that has been used to monitor replication initiation to instead identify those regions of the genome last to complete replication. This approach is based on a combination of flow cytometry and high throughput sequencing. Although this report focuses on the application of this technique to yeast, the approach can be used with any cells that can be sorted in a flow cytometer according to DNA content.
Introduction
Eukaryotic genome replication is initiated at multiple discrete sites, called origins of replication, from which replication forks proceed in both directions (reviewed in Fragkos et al., 20151). Origins vary in both their timing and efficiency of firing, and several techniques have been developed to monitor replication origin activity and elucidate the causes of this variation. The activity of individual origins can be inferred from levels of single-stranded DNA2, which forms around active origins, or by using 2D gel electrophoresis to monitor specific replication intermediates3, both of which can be detected with radioactive probes. Both of these techniques are more easily applied in S. cerevisiae than in mammalian cells, because origins are limited to specific known sequences in the former. With the advent of microarrays, it became possible to assess origin firing globally. This was first done by labeling DNA with heavy isotopes, releasing cells from a G1 block into medium containing light isotopes, and then monitoring the formation of heavy-light hybrid DNA across the genome4. The introduction of high throughput sequencing allowed similar genome-wide monitoring of origin activity without the requirement of expensive isotopic labeling. Cells were sorted in a flow cytometer according to DNA content and their DNA subjected to deep sequencing. Because sequence coverage proceeds from 1x to 2x over the course of S phase, relative replication timing can be assessed by comparing read depths of cells in S phase to those in G1 or G25,6. These techniques, particularly applied to yeast, led to a deeper understanding of how DNA sequence, chromatin structure, and DNA replication proteins regulate origin timing and efficiency.
Faithful transmission of genetic information during cellular proliferation requires not only successful initiation of DNA replication, which takes place at origins, but also successful completion of replication, which occurs where replication forks meet. Like initiation of replication, the timing of completion of replication varies across the genome with certain regions remaining unreplicated even late in the cell cycle. Such regions may be particularly distant from active replication origins or may contain sequences or chromatin structures that impede DNA polymerases. Late replicating regions can manifest themselves as fragile sites, which are associated with chromosomal breakage and higher mutation rates, and have been implicated in cancer and aging7,8,9. However, despite the importance of proper completion of DNA replication in maintenance of genome stability, our understanding of where and how this takes place has lagged far behind that of replication initiation. And while individual genes whose late replication has been associated with disease have been studied with, for example, qPCR10, global studies directed at elucidating the locations and underlying causes of late replication have been lacking. Here we describe a technique we refer to as "G2 seq" in which we combine flow cytometry with high throughput sequencing to shed light on the completion of genome replication in yeast11. With minor changes, this protocol can be adapted to any cells that can be flow-sorted according to DNA content.
Protocol
1. Preparation of Cells for Flow Cytometry Sorting
- Inoculate 15 mL test tubes containing 8 mL each of YEPD broth such that the cultures reach a density of 5 x 106 to 1.5 x 107 cells per mL after overnight growth (see discussion note 1).
- Spin down yeast cells (1,400 x g, at room temperature or 4 °C) in a 15 mL screw cap centrifuge tube for 5 min, resuspend cells in 1.5 mL 70% ethanol, and transfer to a 1.6 mL microfuge tube, letting this sit for 1 h at room temperature or at least 3 h on ice (can be stored indefinitely at 4 °C) (see Discussion point (2)).
- Spin down yeast cells in the microfuge (14,000 x g, 4 °C) for 1 min, resuspend in 1 mL 50 mM sodium citrate, pH 7.2, spin down (14,000 x g, 4 °C) for 1 min, aspirate supernatant, resuspend in 1 mL 50 mM sodium citrate, pH 7.2, containing 0.25 mg/mL RNAse solution for 1 h at 50 °C or overnight at 37 °C in heat block or water bath.
- Add 50 µL proteinase K solution (20 mg/mL resuspended in 10 mM Tris pH 7.5, 2 mM CaCl2, 50% weight to volume glycerol) and incubate for 1 h at 50 °C.
- Spin down cells (14,000 x g, 4 °C), resuspend cells in 1 mL 50 mM sodium citrate, pH 7.2, spin down (14,000 x g, 4 °C), aspirate supernatant with vacuum, resuspend in 1 mL 1 µM Green nucleic acid strain resuspended in 50 mM sodium citrate, pH 7.2 and incubate in the dark for 1 h at room temperature, sonicate 2x for 2 s each (output power 2 watts).
2. Cell Sorting
- Using a cell sorter, sort cells according to DNA content into phases G1, S, "early G2," and "late G2", collecting at least 1.6 million haploid cells from each cell cycle phase as in Figure 1 (see Discussion point (3)).
- Transfer cells to microfuge tubes and spin down cells for 20 min at 14,000 x g at 4 °C in the microfuge and aspirate supernatant with vacuum (the pellet can be stored frozen at -20 °C indefinitely; see discussion point (4)).
3. DNA Extraction and the Preparation of Sequencing Libraries
NOTE: The following steps are based on "protocol I" in the Yeast Genomic DNA extraction Kit (see Table of Materials).
- Add 120 µL of "Digestion Buffer," a proprietary buffer that allows yeast lytic enzyme to digest the cell wall, and 5 µL of yeast lytic enzyme, resuspend by vortexing, and incubate at 37 °C for 40 – 60 min.
- Add 120 µL of "Lysis Buffer," a proprietary buffer that contains detergent, and vortex hard for 10 – 20 s.
- Add 250 µL chloroform – mix thoroughly for 1 min.
- Spin at maximum speed in a microfuge for 2 min.
- Load the supernatant onto the DNA binding column and centrifuge at maximum speed in a microfuge for 1 min.
- Add 300 µL of "DNA Washing Buffer," a proprietary buffer that contains ethanol, and centrifuge for 1 min at maximum speed in a microfuge. Discard the flow through, add 300 µL of DNA Wash Buffer, and repeat the wash process.
- Transfer the spin column to a fresh microfuge tube, add 60 µL of water (not TE) – wait 1 – 3 min and then centrifuge for 10 s to elute the DNA.
- Measure concentration by adding 5 µL of DNA to 195 µL of dsDNA high sensitivity reagent that has been diluted 1:200 in dsDNA high sensitivity buffer and then measure fluorescence in a fluorometer, using 10 µL of supplied standards for calibration; expect between 10 and 50 ng of DNA total yield.
- Sonicate (Peak Incident Power 450, Duty Factor 30%, Cycles per Burst 200, Treatment Time 60 s, Water Level 6) to break up DNA into 250 – 350 bp fragments.
- Prepare a library using DNA sample preparation kit and sequence it (at least 10 million 50 bp single-end reads) on the DNA sequencing instrument in rapid mode (see discussion note (5)).
4. Analysis of Sequencing Data and Generation of Replication Profiles
- Align sequences to Saccharomyces cerevisiae sacCer3 genome using Gsnap12 (see discussion note (6)).
- Determine per-base pair read depths using Bedtools' "genomeCoverageBed" software13.
- Remove artefactual spikes in read depths, if present (see discussion note (7)).
- Smooth read depths by chromosome using medians in a 20 kb sliding window using "runMean" function in R package "caTools" (see discussion note (8)).
- Plot read depths in S phase, early G2, and late G2 as a percentage of completely replicated read depths (see discussion note (9)).
Representative Results
We have used the procedure described above to identify late replicating sites in budding yeast. Testing this approach using a known late replicating region on an artificial chromosome proved the technique to be accurate and reliable. Our results have also demonstrated the biological importance of timely completion of replication by showing that a late replicating region on chromosome 7, which we identified as late replicating on the basis of our G2-seq results, was lost approximately three-fold more frequently than a comparable control region11.
A specific example of results with G2-seq is shown in Figure 2. We had hypothesized that there would be regions of the genome that replicated particularly late in cells lacking a protein deacetylase called Sir2. Figure 2A shows the progression of data analysis for wild type (blue) and sir2 (red). The raw data (upper rows) contain large spikes, most of which are due to the presence of Ty elements. These spikes are removed by capping read depths at 2.5x the median read depth for each sample (middle rows). The despiked data are then smoothed with a 20 kb sliding window (bottom row). Finally, data are normalized and plotted as ratios to the levels in G1. A typical late-replicating region that appears in the sir2 mutant is highlighted in Figure 2C.
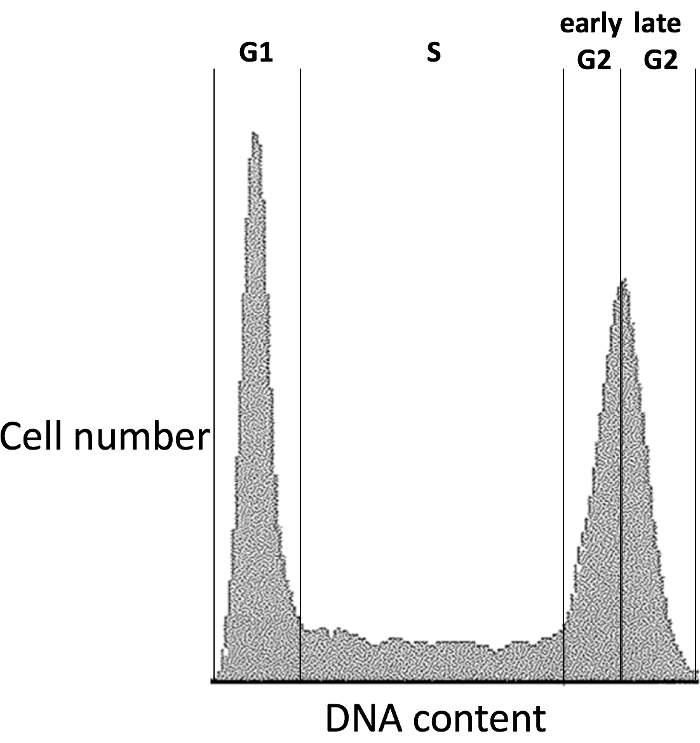
Figure 1. Flow cytometry profile indicating cell cycle fractions used; outer limits of all gates marked. Please click here to view a larger version of this figure.
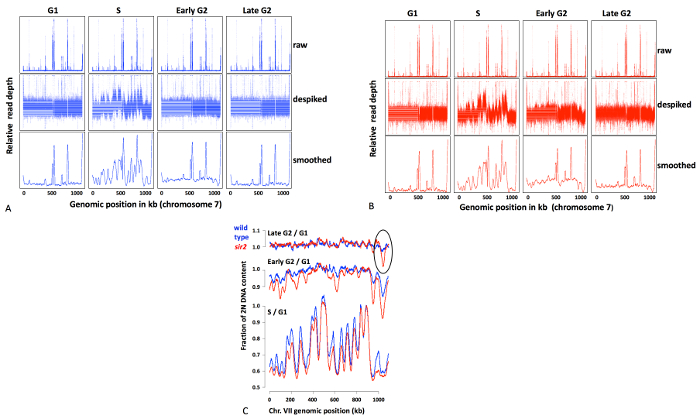
Figure 2. (A-B) Progression of data processing from raw data (top rows), despiked data (middle rows) and smoothed data (bottom rows) for wild type (A) and sir2 (B). (C) Ratios of S phase-, early G2-, and late G2-to G1 read depths. Wild type in blue and sir2 in red. The highlighted region in the upper right shows a late replicating region specific to sir2 mutant. Scale indicates fraction of complete (2N) replication; a completely unreplicated cell would appear at 0.5, a completely replicated cell at 1. Please click here to view a larger version of this figure.
Discussion
While this technique is robust and relatively straight forward, particular attention should be devoted to the following:
(1) We recommend that cultures grow for at least 12 h before they reach log phase, since differences manifest in cell cycle distributions if cultures are harvested after having reached the desired density just 4 h after inoculation. Our assumption is that a cell cycle distribution that has reached a relatively stable equilibrium better represents a "real" log phase distribution than a distribution that is still in flux when a saturated culture has been transferred more recently to fresh medium.
(2) Instead of spinning down the cells before fixing, cells can be also fixed by directly adding 100% ethanol to the yeast culture so that the final concentration of ethanol is 70%. Cells can be grown in either synthetic or rich (YEPD) medium.
(3) Cell cycle phases are visualized for sorting by plotting cell number as a function of 530 nm emission area. We have found that the inflection points between G1 and S and between S and G2 serve as robust landmarks for delineating S phase, and we use the G2 maximum as the boundary to separate early and late G2. We use the terms "early G2" and "late G2" for clarity and brevity, perhaps at the expense of accuracy in the sense that cells are technically in S phase until they have completed DNA replication. Although the goal of G2-seq is to identify very late-replicating sites, which are expected to be revealed in the late G2 fraction, we collect S phase and early G2 cells to follow replication through all phases of the cell cycle.
(4) It is critically important that cells be spun down promptly after sorting. Do not let cells sit overnight before spinning them down. We have had problems recovering cells by centrifugation if they have been stored at 4 °C, suggesting that they become fragile upon sorting and lyse with time.
(5) As few as 5 million reads is sufficient, but plots become less smooth with lower numbers of reads. Data sets with even fewer reads may be able to be accommodated by smoothing with windows larger than 20 kb, but we have not tried this.
(6) Any widely-used alignment software (e.g., Bowtie14, Novalign (http://www.novocraft.com), BWA15, or Gsnap12) will work.
(7) High throughput sequencing data from yeast show occasional spikes in read depths, which often reflect Ty elements. We eliminate these by substituting 2.5x median sample read depths for read depths outside of regions known to contain repetitive stretches (e.g., the rDNA) that are greater than 2.5x median. The rDNA and the ends of chromosomes are excluded because they are known to contain repetitive elements. An example of this "despiking" using the R language is the following:
# create a data frame called "reads" containing data:
chr <- rep('chrI', 10)
pos <- seq(1, 10)
rd <- c(3, 4, 6, 5, 88, 2, 9, 10, 900, 1)
reads <- data.frame('chromosome' = chr,
'position' = pos,
'read_depth' = rd)
# "despike" the read depths:
reads[,'despiked'] <- ifelse(reads[,3] > (2.5 * median(reads[,3])),
(2.5 * median(reads[,3])),
reads[,3])
"Capping" or "despiking" can also be achieved by excluding reads that map to more than one location, but this will eliminate reads that map to, for example, just two locations. Instances of reads that map to more than one location, but still provide valuable information, are not uncommon when working with S. cerevisiae, whose genome resulted from an ancestral duplication16.
(8) We have obtained comparable results with both the "rollmedian" function in the R package "zoo" (https://cran.r-project.org/web/packages/zoo/index.html) and the "runmean" function in the R package "caTools" (https://cran.r-project.org/web/packages/caTools/index.html).
(9) We have experimented with numerous methods to normalize read depths, all of which gave comparable results. Our favored approach is based on the assumption that at least some regions of the genome are fully replicated in our S phase sample. We determined the read depth that represented full replication as the median read depth in our data set at sites corresponding to the 12 earliest firing high confidence ("confidence" score >= 7) origins in a publicly available data set. (http://cerevisiae.oridb.org/data_output.php?main=sc_ori_studies&table=Raghu2001_ori&ext_format=&format=tab). For most applications, the approach taken for normalization is not critical and there are many reasonable options, most simply just normalizing to read count totals. However, it is worth noting that if there are large differences in the numbers of repetitive sequences between samples, normalization to read count totals can be misleading. For example, when comparing two strains whose rDNA arrays differ by several fold, normalizing to total read counts can make certain regions of the genome of the strain with the larger rDNA array artefactually appear to replicate later than the strain with the smaller array.
In summary, G2-seq represents a logical extension of existing techniques for replication profiling. Like other sequencing- and microarray-based techniques, G2-seq has advantages over 2D gel analysis of genome wide coverage and the lack of requirement of knowledge of the location of replication origins. The latter is particularly relevant in mammalian cells where, unlike in budding yeast, replication origins are not limited to specific sequences. On the other hand, 2D gels provide a level of resolution of molecular intermediates in DNA replication completely unattainable by G2-seq. Extending G2-seq to other organisms whose cells can be sorted according to DNA content should be relatively straight forward, however, the highly repetitive nature of the genomes of higher eukaryotes makes alignment of short read sequences much more challenging. Emerging sequencing technologies that provide significantly longer read length may prove useful in this regard, especially since G2-seq should be relatively robust to the higher error rates that can be associated with longer reads17. In our experience, the technical problem that is most likely to cause problems is letting cells sit after they have been sorted and prior to isolation of DNA. These cells appear to be particularly fragile and to lyse with time, and DNA should be isolated within at most an hour or two of sorting. Promising future directions for this technique include dividing both S and G2 phases into increasing numbers of fractions to obtain higher resolution of the dynamics of genome replication, from early initiation to late completion.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
This work was supported by grant NIH GM117446 to A.B.
Materials
| YeaStar Genomic DNA kit | Genesee Scientific | 11-323 | |
| 1 µM SYTOX Green | ThermoFisher | 57020 | resuspend in 50 mM sodium citrate, pH 7.2 |
| 50 mM sodium citrate, pH 7.2 | |||
| RNase solution (0.25 mg / mL) | Sigma | R6513 | 0.25 mg / mL RNaseA resuspended in 50 mM sodium citrate, pH 7.2, boil RNase solution for 10 minutes before the first use only, and from then on store at -20° |
| proteinase K solution (20 mg / mL) | ThermoFisher / Invitrogen | 25530-015 | resuspend in 10 mM Tris, pH 7.5, 2 mM CaCl2, 50% glycerol, store at -20°C |
| Model 50 Sonic Dismembrator | Fisher Scientific | FB50A220 | |
| BD Biosciences FACSAria II | BD Biosciences | 644832 | |
| Zymo-spin III columns | Zymo Research | C1005 | |
| Qubit dsDNA HS Assay Kit | ThermoFisher | Q32851 | |
| Qubit 3.0 Fluorometer | ThermoFisher | Q33216 | |
| Covaris Model LE220 Focused-Ultrasonicator | Covaris | 500219 | |
| Illumina TruSeq DNA LT Sample Prep Kit | Illumina | 15026486 | |
| Illumina HiSeq 2500 instrument | Illumina | SY–401–2501 | |
| gsnap alignment software | open source software / Genentech | http://research-pub.gene.com/gmap |
Riferimenti
- Fragkos, M., Ganier, O., Coulombe, P., Mechali, M. DNA replication origin activation in space and time. Nat Rev Mol Cell Biol. 16, 360-374 (2015).
- Santocanale, C., Diffley, J. F. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 395, 615-618 (1998).
- Brewer, B. J., Fangman, W. L. A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell. 55, 637-643 (1988).
- Raghuraman, M. K., et al. Replication dynamics of the yeast genome. Science. 294, 115-121 (2001).
- Muller, C. A., Nieduszynski, C. A. Conservation of replication timing reveals global and local regulation of replication origin activity. Genome Res. 22, 1953-1962 (2012).
- Koren, A., Soifer, I., Barkai, N. MRC1-dependent scaling of the budding yeast DNA replication timing program. Genome Res. 20, 781-790 (2010).
- Lang, G. I., Murray, A. W. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol Evol. 3, 799-811 (2011).
- Durkin, S. G., Glover, T. W. Chromosome fragile sites. Annu Rev Genet. 41, 169-192 (2007).
- Dillon, L. W., Burrow, A. A., Wang, Y. H. DNA instability at chromosomal fragile sites in cancer. Curr Genomics. 11, 326-337 (2010).
- Widrow, R. J., Hansen, R. S., Kawame, H., Gartler, S. M., Laird, C. D. Very late DNA replication in the human cell cycle. Proc Natl Acad Sci U S A. 95, 11246-11250 (1998).
- Foss, E. J., et al. SIR2 suppresses replication gaps and genome instability by balancing replication between repetitive and unique sequences. Proc Natl Acad Sci U S A. 114, 552-557 (2017).
- Wu, T. D., Reeder, J., Lawrence, M., Becker, G., Brauer, M. J. GMAP and GSNAP for Genomic Sequence Alignment: Enhancements to Speed, Accuracy, and Functionality. Methods Mol Biol. 1418, 283-334 (2016).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics. , 17 (2010).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754-1760 (2009).
- Kellis, M., Birren, B. W., Lander, E. S. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 428, 617-624 (2004).
- Goodwin, S., McPherson, J. D., McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 17, 333-351 (2016).
