The photoactivation and imaging approach allows for observation and facile quantification of rapid, polarized signaling responses. To illustrate its capabilities, reproduced here is an experiment examining the spatiotemporal correlation between TCR-induced DAG accumulation and centrosome reorientation. 5C.C7 T cell blasts were retrovirally transduced with two fluorescent reporters: a DAG biosensor containing the tandem C1 domains from protein kinase C-θ linked to GFP (C1-GFP) and RFP-tubulin to monitor the centrosome. The T cells were then attached to chambered coverglass containing photoactivatable MCC- I-Ek. C1-GFP was imaged using TIRF microscopy and RFP-tubulin in epifluorescence mode. As shown in Figure 2, localized photoactivation of the surface beneath the T cell induces the accumulation of DAG in the irradiated zone. This is followed within seconds by the reorientation of the centrosome to the same region.
C1-GFP accumulation can be quantified by calculating the normalized fluorescence intensity, after background correction, at the center of the photoactivated region over time (Figure 3). Centrosome reorientation in response to photoactivation can be quantified by calculating the distance between the centrosome and the center of the photoactivated region as a function of time (Figure 4).
This method can be used to examine a wide array of rapid, polarized signaling responses, essentially anything that can be monitored using a fluorescent probe. For example, TCR photoactivation is utilized to monitor early signaling microcluster formation using fluorescently labeled Grb2 (Grb2-GFP) (Figure 5). Similar to the DAG accumulation and centrosome reorientation responses, Grb2-GFP polarization occurs shortly following photoactivation (Figure 5), and can be quantified using the normalized fluorescence intensity approach.
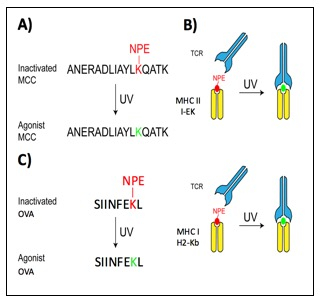
Figure 1: Photoactivation system using photocaged MCC peptide.
(A) Photocaged strategy for MCC peptide. A NPE group is attached to the lysine residue of the MCC peptide. UV irradiation results in the removal of NPE, restoring MCC to its native form. (B) When NPE is bound to MCC, the 5C.C7 TCR cannot bind to MCC. Removal of NPE results in recognition of the 5C.C7 TCR to the native MCC peptide. (C) The photocaged strategy has been adapted to work with CD8+ T cells, in which photocaged OVA peptide is used to activate the OT-1 TCR upon UV irradiation.
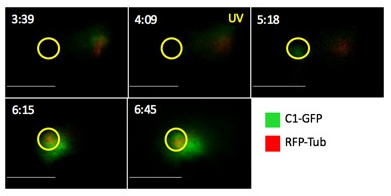
Figure 2: Time-lapse montage of DAG accumulation and centrosome reorientation following photoactivation of TCR in 5C.C7 T cell.
5C.C7 cell expressing a DAG biosensor, containing tandem C1-domains of PKC- fused to GFP (C1-GFP) and Tag-RFP Tubulin (RFP-Tub), a fluorescent reporter for the centrosome. The montages illustrate the polarized response of 5C.C7 T cells over time. The yellow oval and text indicate the time and location of photoactivation. Over time, C1-GFP accumulates at the photoactivated region, followed by centrosome reorientation towards the photoactivated region. Scale bar: 10 µm.
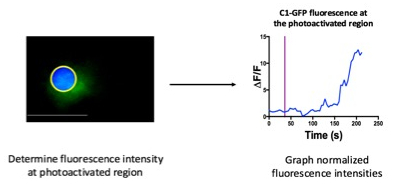
Figure 3: Quantification of DAG accumulation at the photoactivated region
DAG, C1-GFP, accumulation at the photoactivated region (left) can be quantified by calculating the normalized fluorescence intensity, after background correction, at the photoactivated region (yellow circle with blue shading) over time. Data are normalized relative to the nine frames obtained just before photoactivation. C1-GFP enrichment at the photoactivated region can be calculated by the division of the mean fluorescence intensity in the irradiated region by the mean fluorescence intensity of the entire cell, following background correction. The equation: ΔF/F = ((FI-FIb)/mean(FI1-9)-FIb)), in which FI is the mean fluorescence intensity, can be used to calculate the normalized fluorescence intensity. The normalized fluorescence intensity at the photoactivated region can then be graphed as a function of time (right). Purple line indicates the time of photoactivation. Scale bar: 10 µm.
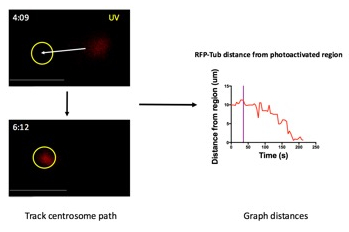
Figure 4: Quantification of centrosome reorientation.
Determine the coordinates of the centrosome (RFP-Tub) over time by manually tracking the centrosome path toward the photoactivated region (white arrow) at each time point (left). Determine the coordinates of the center of the photoactivated region (yellow circle). The distance between the centrosome and the center of the photoactivated region can be calculated at each time point using the equation for distance = √((x2-x1)2+(y2-y1)2), in which x2 and y2 are the x and y coordinates for the center of the area of the centrosome, and x1 and y1 are the x and y coordinates of the center of the photoactivated region. The distance of the centrosome from the center of the photoactivated region at each time point can then be graphed as a function of time (right). Purple line indicates the time of photoactivation. Scale bar: 10 µm.
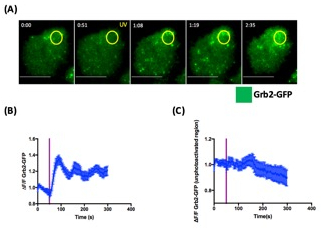
Figure 5: Grb2-GFP microcluster formation.
(A) Representative 5C.C7 cell expressing fluorescently labeled Grb2 (Grb2-GFP). The montages illustrate the polarized response of Grb2-GFP overtime. The yellow oval and text indicate the time and location of photoactivation. Over time, Grb2-GFP accumulates at the photoactivated region. (B) Quantification of Grb2-GFP fluorescence intensity at the photoactivated region over time. N = 15 cells. (C) Quantification of Grb2-GFP fluorescence intensity in a control region outside of the photoactivated zone. N = 15 cells.
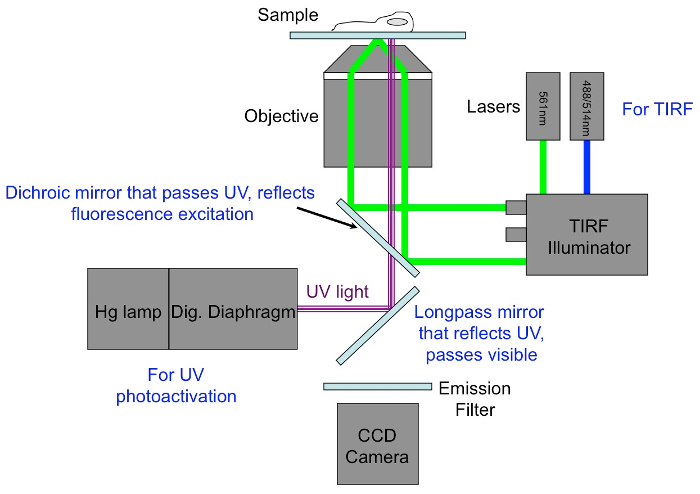
Figure 6: Microscope configuration for photoactivation and TIRF imaging.
488 nm and 561 nm lasers are used for TIRF and epifluorescence imaging of green and red probes, respectively. UV light for photoactivation is taken from a mercury (Hg) lamp attached to a digital diaphragm system capable of illuminating small, user-defined regions. Light from the Hg lamp/diaphragm is reflected onto the sample by a longpass mirror positioned beneath the dichroic mirror that reflects the imaging lasers. The dichroic mirror must be designed to pass UV light. Please click here to view a larger version of this figure.
