Assessing the Viability of a Synthetic Bacterial Consortium on the In Vitro Gut Host-microbe Interface
Summary
Gut host-microbe interactions were assessed using a novel approach combining a synthetic oral community, in vitro gastrointestinal digestion, and a model of the small intestine epithelium. We present a method that can be adapted to evaluate cell invasion of pathogens and multi-species biofilms, or even to test probiotic formulations' survivability.
Abstract
The interplay between host and microbiota has been long recognized and extensively described. The mouth is similar to other sections of the gastrointestinal tract, as resident microbiota occurs and prevents colonisation by exogenous bacteria. Indeed, more than 600 species of bacteria are found in the oral cavity, and a single individual may carry around 100 different at any time. Oral bacteria possess the ability to adhere to the various niches in the oral ecosystem, thus becoming integrated within the resident microbial communities, and favouring growth and survival. However, the flow of bacteria into the gut during swallowing has been proposed to disturb the balance of the gut microbiota. In fact, oral administration of P. gingivalis shifted bacterial composition in the ileal microflora. We used a synthetic community as a simplified representation of the natural oral ecosystem, to elucidate the survival and viability of oral bacteria subjected to simulated gastrointestinal transit conditions. Fourteen species were selected, subjected to in vitro salivary, gastric, and intestinal digestion processes, and presented to a multicompartment cell model containing Caco-2 and HT29-MTX cells to simulate the gut mucosal epithelium. This model served to unravel the impact of swallowed bacteria on cells involved in the enterohepatic circulation. Using synthetic communities allows for controllability and reproducibility. Thus, this methodology can be adapted to assess pathogen viability and subsequent inflammation-associated changes, colonization capacity of probiotic mixtures, and ultimately, potential bacterial impact on the presystemic circulation.
Introduction
Humans cohabit with bacteria, which are present at the same number as human cells1. Hence, it is of crucial important to obtain a comprehensive understanding of the human microbiome. The oral cavity is a unique environment in that it is divided into several smaller habitats, thus containing a large variety of bacteria and biofilms in those different locations. Being an open ecosystem, some species in the mouth may be transient visitors. However, certain microorganisms colonize soon after birth and form organized biofilms2. These are found in the teeth surface above the gingival crevice, the subgingival crevice, tongue, mucosal surfaces and dental prosthetics and fillings3. Bacteria can also be present as flocs and planktonic cells in the lumen of the tooth canal, either intermixed with necrotic pulp tissue or suspended in a fluid phase.
There is active, continuous cross-talk between host cells and the resident microbiota4. Bacteria communicate within and between species, and only a small proportion of the natural colonizers can adhere to tissues, while other bacteria attach to these primary colonizers. For instance, cell-cell binding between microorganisms is key for integrating secondary colonizers into oral biofilms, and building complex networks of interacting microbial cells4. Around 70% of bacterial aggregates in a saliva sample are formed by Porphyromonas sp., Streptococcus sp., Prevotella sp., Veillonella sp., and unidentified Bacteroidetes. F. nucleatum is an intermediate colonizer in the subgingival biofilm and aggregates with the late colonizers P. gingivalis, T. denticola, and Tannerella forsythia, which are implicated in periodontitis5. In addition, Streptococcus mitis occupies both mucosal and dental habitats, while S. sanguinis and S. gordonii prefer to colonize teeth3. Thus, S. sanguinis is present in lower incisors and canines, while Actinomyces naeslundii has been found in upper anteriors6.
In addition, the indigenous microbiome plays a role in maintaining human health2. Resident microbiota participates in immune education and in preventing pathogen expansion. This colonisation resistance occurs because the native bacteria may be better adapted at attaching to surfaces, and more efficient at metabolising the available nutrients for growth. Although probiotic strains survive the gastrointestinal passage and remain active, the persistence of autochthonous bacteria swallowed from an upper location of the gastrointestinal tract has not been fully described. Thus, we subjected an artificial community, representative of the oral ecosystem, to simulated gastrointestinal transit conditions. Viability of bacterial cells was assessed using a multicompartment model resembling the gut epithelium. Current gut simulators offer suitable reproducibility in terms of analysis of the luminal microbial community7. However, bacterial adhesion and host-microbe interaction are separately addressed, as combining cell lines with microbial communities is challenging8. In contrast, we present a framework that provides potential mechanistic explanation of successful colonization events reported on the gut interface. Indeed, this model can be jointly used with a static gut model to evaluate the impact of microbial communities on host surface signalling.
Protocol
1. Strains and Culture Conditions
NOTE: The synthetic oral community was composed by strains commonly present in the oral microbiome3.
- Obtain the following strains from the American Type Culture Collection (ATCC): Aggregatibacter actinomycetemcomitans (ATCC 43718), Fusobacterium nucleatum (ATCC 10953), Porphyromonas gingivalis (ATCC 33277), Prevotella intermedia (ATCC 25611), Streptococcus mutans (ATCC 25175), Streptococcus sobrinus (ATCC 33478), Actinomyces naeslundii (ATCC 51655), Streptococcus gordonii (ATCC 49818), Actinomyces viscosus (ATCC 15987), and Streptococcus mitis (ATCC 49456).
- Acquire Veillonella parvula from the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures (DSM 2007), Streptococcus sanguinis from the BCCM/LMG Bacteria Collection (LMG 14657) and use Streptococcus salivarius strain TOVE-R9, and Streptococcus oralis10.
2. Development of a Multispecies Community Representative of the Oral Microbiome
NOTE: Generate a synthetic community to simulate the potential adhesion capacity of oral bacteria to the in vitro gut epithelium. Grow bacteria on blood agar plates supplemented with 5 μg/mL hemin, 1 μg/mL menadione, and 5% sterile defibrinated horse blood, as described in Hernandez-Sanabria et al. (2017)11. In brief:
- Dissolve 100 mg of hemin in 2 mL of 1 M NaOH, add 100 mL of distilled autoclaved water. Store in a dark container. Sterilize through a 0.22 µm filter before adding to medium.
- Dissolve 100 mg of menadione in 20 mL of 96% ethanol. Sterilize through a 0.22 µm filter before adding to medium.
- Prepare 1 L of blood agar medium (see Table of Materials) according to the manufacturer’s instructions and autoclave. Let cool down before adding the horse blood and the supplements. Mix and pour the plates.
- Prepare modified Brain Heart Infusion (BHI) broth as previously reported12. Add 5 μg/mL of hemin and 1 μg/mL of menadione after autoclaving.
- Dispense 9 mL of the medium in Hungate tubes, and close with a rubber stopper and an aluminium cap. Flush the tubes with N2/CO2 (90%/10%).
- Retrieve 1 mL of anaerobic medium with a syringe and place it in a 1.5 mL microcentrifuge tube. Pick single colonies of each bacterial species, resuspend in the medium, and transfer back into the anaerobic modified BHI. Incubate for 48 h at 37 °C, except for S. salivarius, which must be incubated for 24 h.
- If needed, confirm purity of the cultures prior to assembling the synthetic community. In brief:
- Extract DNA from the liquid cultures as in Hernandez-Sanabria et al. (2010)13 and amplify the 16S rRNA gene using the primers 27F (AGAGTTTGATYMTGGCTCAG) and 1492R (TACGGYTACCTTGTTACGACT), and the following program: 5 minutes at 95 °C, 30 cycles of 95 °C for 1 min, 55 °C for 1 min, and 72 °C for 1.5 min, followed by a final extension step of 5 min at 72 °C.
- Purify the PCR product with a commercial kit (see Table of Materials), following the manufacturer’s instructions.
- Perform the sequencing reaction of the purified products in a 10 μL total volume containing 0.5 μL of dye (see Table of Materials), 3.2 pmol of M13f primer (CGCCAGGGTTTTCCCAGTCACGAC), 2.0 μL of 5x sequencing buffer, and 20 ng of template, using a commercial sequencing system with kit (see Table of Materials).
NOTE: We had this procedure performed commercially. - Perform a BLAST search on all the sequences (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to determine the closest known taxon and compare with the sequence of the original strain using the RDP Classifier online tool (http://rdp.cme.msu.edu/) and the SINA Alignment service (https://www.arb-silva.de/aligner/).
- Measure the cell number at the end of the incubation, using flow cytometry and SYBR Green/Propidium Iodide stain as described in Hernandez-Sanabria et al. (2017)11. Dilute the cultures to 105 cells mL-1, using modified BHI.
- Add 1 mL of S. mitis into 75 mL of anaerobic BHI and incubate until stationary phase (6 h). Following, collect 0.75 mL of each diluted strain and mix under anaerobic conditions.
- Once all the cultures have been mixed, take 1 mL of the microcosm, and add to the 75 mL of BHI. Incubate the synthetic community under 10% CO2, 10% H2, 80% N2 for 48 h at 37 °C and 200 rpm (see Table of Materials).
- Measure cell density as in 2.8. before presenting the community to the cell model. Composition of the community can be assessed with quantitative Real-Time PCR, using the primers and annealing temperatures in Table 1 of Slomka et al. (2017)10. In addition, use 16S rRNA gene amplicon sequencing to provide information on the initial members present13,14. For either identity-confirming protocol, use cDNA as a template to indicate the bacteria actively transcribing proteins, and use DNA as a template to reveal presence/absence of the strains.
3. General Cell Culture Practices
NOTE: For general aspects of cell culture, the authors refer to Master and Stacey (2007)15. Obtain cell lines used from the European Collection of Authenticated Cell Cultures (Caco-2 ECACC 86010202 and HT29-MTX-E12 ECACC 12040401, Public Health England, UK). Reagents for cell culture can be purchased (see Table of Materials), unless otherwise specified.
- Maintenance and passaging of Caco-2 and HT29-MTX cell lines
NOTE: Caco-2 and HT29-MTX cells are routinely grown in 25 cm2 tissue culture flasks containing supplemented DMEM medium (Table 1). Cells are trypsinized when 60–70% confluence is reached.- Remove the cell culture medium from the flasks. Wash twice with 5 mL of PBS without Ca++/Mg++.
- Add 2 mL of a 0.5 mg/L solution of trypsin and 0.22 g/L ethylene diamine tetraacetic acid (EDTA). Distribute the trypsin solution by gently moving the flask. Remove 1.5 mL of the trypsin solution and incubate the flask at 37 °C for 10 min.
- Check under the microscope if cells are detached from the flask surface. Incubate for additional 5 min, if needed.
- Add 5 mL of supplemented cell culture media to the flask to inactivate the trypsin. Pipet up and down to obtain a homogenous single cell suspension.
- Immediately, take a 50 µL aliquot of the cell suspension and mix with sterile 0.4% Trypan blue solution in a 1:1 (v/v) proportion for Caco-2 cells or 1:5 v/v for HT29-MTX. Mix by pipetting and load into a counting chamber (see Table of Materials).
- Count the viable cells (white bright cells) under the microscope (see manufacturer’s instructions).
- Seed in a new flask at a density of 4 x 105 cells/cm2 and 1 x 104 cells/cm2, for Caco-2 and HT29-MTX, respectively.
NOTE: (1) Caco-2 cells differentiate and form tight junctions once reaching confluency. It is extremely important to avoid over-confluency before trypsinization. Overgrowth of cells in flask can cause failure in cell detachment after trypsinization and/or cell clumps. (2) HT29-MTX cells are mucus-producing cells with high metabolic activity16. These features may cause a fast acidification of the cell culture media, especially if cells are at high density. HEPES buffer can be added to the cell culture medium (Table 1) to maintain the pH between 7 and 7.2. If the pH drops, medium can be exchanged when confluency is less than 50%. However, if confluency >50%, the cell culture must be split.
4. Assembly of a Multicompartment Cell Model Simulating the Gut Host-microbe Interface
NOTE: Complete the co-culture in double chamber wells (diameter 24 mm, pore size 0.4 μm; see Table of Materials).
- Add 1.5 mL of complete cell culture media to the apical compartment and 2 mL to the basal. Pre-incubate for 20–30 min to obtain even distribution of the cell suspension when seeding.
- Trypsinize the cell culture of Caco-2 and HT29-MTX as described in section 3. The trypsinization procedure must be accurately followed to achieve homogenous distribution of both cell lines, in a single cell suspension, without clumps.
- Count the viable cells as described in 3.1.8.
- Adjust the cell density to 6.5 x 104 cells/cm2 in a 90/10 proportion of Caco-2/HT29-MTX cells.
- Homogenize the cell suspensions and add the corresponding volume of Caco-2 and HT29-MTX cells to a pre-filled sterile container with supplemented cell culture media. Remove the pre-incubated media from the apical side of the chamber by aspiration.
- Gently mix the cell suspension by pipetting, and transfer 1.5 mL to the apical compartment of the chamber inserts. The seeding procedure requires a constant homogenization of the cell suspension, as cells tend to sediment. Depending on the experience of the user and working speed, cell suspension must be mixed after dispensing 1–3 wells.
- Move plates gently backward and forward and then right to left to right (5–10 times) to obtain an even spread of cells in the well. Transfer to incubator and repeat the movement of the plates.
- Maintain the co-culture of Caco-2/HT29-MTX cells for 15 days, refreshing apical and basal compartments every 2 days with supplemented DMEM. After this time, the cell culture media can be changed to supplemented DMEM without antibiotic/antimycotic solution.
- Maintain the system until 20–21 days post-seeding by refreshing the medium every 2 days.
- Measure the epithelial barrier integrity before starting the assay, using an Electrical Resistance System (see Table of Materials), following the manufacturer instructions.
- Ensure that TEER equipment is fully charged (>24 h) before starting the measurements.
- Disconnect the TEER equipment from the power supply and cover with a plastic autoclave bag. Make a hole in the bag to introduce the electrode and plug into the input port on the meter. Spray with 70% ethanol/water (v/v) before introducing the TEER equipment into the flow cabinet.
- To disinfect the electrode, immerse the electrode tips in 70% ethanol/water (v/v) solution for 15 min. Allow to air dry for 15 s. Rinse the electrode in pre-warmed cell culture media.
- Take the cells out of the incubator and allow cells to come to room temperature inside the flow cabinet.
- Make sure that the meter is disconnected from the charger. Set the mode switch to Ohms and turn the power switch On.
- Measure the cell resistance by immersing the electrode with the shorter tip in the insert and the longer tip out of the well. Keep the electrode at a 90° angle to the plate insert.
5. Bacterial Survival Following In Vitro Gastrointestinal Transit Conditions
NOTE: Prepare simulated digestion fluids following the protocol of Minekus et al. (2014)17, with the modifications described below. Prepare all the dilutions with ultrapure water. Filter-sterilize all solutions through a 0.22 µm filter and perform the digestion procedure under sterile conditions.
- Oral digestion:
- Place 3 mL of the bacterial community in a 50 mL sterile tube, mix with 3 mL of simulated saliva fluid (SSF), containing (in mmol L-1): KCl, 15.1; KH2PO4, 3.7; NaHCO3, 6.8; MgCl2(H2O)6, 0.5, (NH4) CO3, 0.06; HCl, 1.1; CaCl2(H2O)2, 0.75. Add 75 U/mL of α-amylase from human saliva Type IX-A and 2 g/L mucin from porcine stomach type II to the SSF.
- Incubate the mixture for 2 min, at 37 °C, and 100 rpm. Collect a 2 mL sample for DNA extraction and flow cytometry quantification (S1).
- Gastric digestion:
- Add 4 mL of simulated gastric fluid (SGF), containing (in mmol L-1): KCl, 6.9; KH2PO4, 0.9; NaHCO3, 25; NaCl, 47.2; MgCl2(H2O)6, 0.1, (NH4) CO3, 0.5; HCl, 15.6; CaCl2(H2O)2, 0.075. In addition, the SGF contained 2 g/L of mucin from porcine stomach type II.
- Measure the pH and adjust to pH 3 with 1 M HCl, if needed. Incubate at 37 °C and 100 rpm. Collect a 2 mL sample (S2).
- Small intestine digestion:
- Add 4 mL of simulated intestinal fluid (SIF) containing (in mmol L-1): KCl, 6.8; KH2PO4, 0.8; NaHCO3, 85; NaCl, 38.4; MgCl2(H2O)6, 0.33; HCl, 8.4; CaCl2(H2O)2, 0.3; to the digestion tube.
- Adjust to pH 7 with 1 M NaOH, if needed. Incubate at 37 °C and 100 rpm. Collect a 2 mL sample (S3).
6. Bacterial Colonization Ability Following In Vitro Gastrointestinal Transit Conditions
- Centrifuge 2 mL of the small intestine digestion, for 10 min at 2,650 x g.
- Remove the supernatant and mix the bacterial pellet with 5 mL of supplemented DMEM without antibiotic and antimycotic.
- Stain and quantify intact/damaged cells using a flow cytometer (see Table of Materials), as described by Hernandez-Sanabria et al. (2017)11.
- Adjust the cell density to 105 viable cells/mL by diluting in supplemented DMEM without antibiotic and antimycotic.
- Remove the apical medium of the transwell system and add 1.5 mL of the bacterial suspension. Co-culture the system for 2 h under general cell culture conditions (37 °C, 95% humidity, 5% CO2).
NOTE: This time can be extended up to 48 h, depending on the experimental protocol required. The co-culture can be maintained in a different incubator than that used for routine cell maintenance, to avoid contaminations. - Measure the TEER values after incubation, to evaluate the integrity of the epithelial barrier as described in 4.11.
- Upon completing incubation time, remove apical media, and collect 0.5 mL for further analyses (S4). A subsample of media can be used for assessment of cell viability by Lactate Dehydrogenase (LDH) assay (see Table of Materials), following the manufacturer instructions.
- Add 0.5 mL of 10 mM N-acetylcysteine in HBSS (NAC-buffer) to solubilize the mucus produced by the HT29-MTX and to recover adhered bacteria. Incubate the plates at 37 °C for 1 h, under agitation (135 rpm, see Table of Materials).
- Recover the NAC-HBSS in a microcentrifuge tube (S5) and wash twice the cells with 1 mL DPBS.
- Add 0.5 mL of 0.5% Triton X-100 (w/v) on top of the monolayers, disrupt the cells by pipetting, recover the liquid and vortex for 1 min. Speed is crucial to avoid damaging the bacterial cells with the detergent.
- Centrifuge the cells for 5 min at 2,650 x g, and separate the supernatant (S6) and the pellet (S7).
7. Sample Analysis
NOTE: Analyse collected samples (S1-S7) to evaluate bacterial viability and adhesion potential to the intestinal cells, following a simulated gastrointestinal digestion.
- Bacterial viability: Pass samples S1-S5 twice through a 0.45 µm and quantify bacterial cells using live/dead cell staining and flow cytometry, as indicated in step 2.811.
- Adhesion potential: Prepare serial dilutions (-1 to -8) for S1-S6 and streak 10 µL in blood agar and modified BHI plates. Incubate at 37 °C under anaerobic conditions for 24–48 h. Plate the same volume of undiluted S7.
- Taxonomical identification of adhered bacteria:
- Pick individual colonies with a culture loop and resuspend each on 10 µL of nuclease-free water. Collect 4 µL of this suspension and use it as a template for PCR amplification of the 16S rRNA gene using the primers and conditions described in 2.7.1.
- Perform sequencing following the protocol in 2.7.3, and validation of the sequence using the procedure included in 2.7.4.
- Taxonomical identification of adhered bacteria:
- Do further downstream analysis such as total DNA extraction, qPCR quantification and/or 16S rRNA amplicon sequencing, RNA extraction, and transcriptomic profiling as desired.
NOTE: Flow cytometry quantification was performed immediately after sampling. As a control for membrane-permeabilized cells, a sample exposed to 70 °C for 3 min was used. Background noise was assessed by measuring with flow cytometry the digestion fluids or samples obtained from the cell culture model without bacteria. Every sample was measured in triplicate.
Representative Results
This protocol leads to the generation of a model suitable for elucidating the survival and viability of oral bacteria subjected to simulated gastrointestinal transit conditions. The counts of intact cells from individual strains is approximately 108 cells mL-1 prior the creation of the synthetic community, while the multispecies microcosm contained above 90% of viable cells during the establishment of the community (Figure 1A and 1B). Based on the Live/Dead quantification, bacterial viability decreases after each digestion step (Figure 2). This can be a result of acid pH during the gastric passage and of the bile salts contained in the small intestine digestion fluids, as occurring under physiological conditions. Viability during the transit through both in vitro and in vivo gastrointestinal digestion may depend on variations in the environment (e.g., fed vs. fasted conditions), or even on bacteria protection processes (spore formation or enteric coatings).
Flow cytometric counts must be compared with suitable controls, such as heat-killed bacteria or background samples, to perform accurate viable cell quantification18. The harsh conditions of the stomach and small intestine digestion can switch bacteria to viable but non-culturable cells, which are live bacteria that do not either grow or divide. For this reason, plate counts differ from viable cell counts obtained with flow cytometry. For instance, in Figure 3, viable cells recovered from the mucosal interface are coloured in blue in the flow cytometry plot. However, no growth was observed when these mucus samples were plated (results not shown).
The fraction on which bacteria show higher viability rates is the cellular debris (S7), as revealed by plate counting (Figure 4). The number of colonies may be variable, but presence of metabolically active bacteria is found in the less diluted samples.
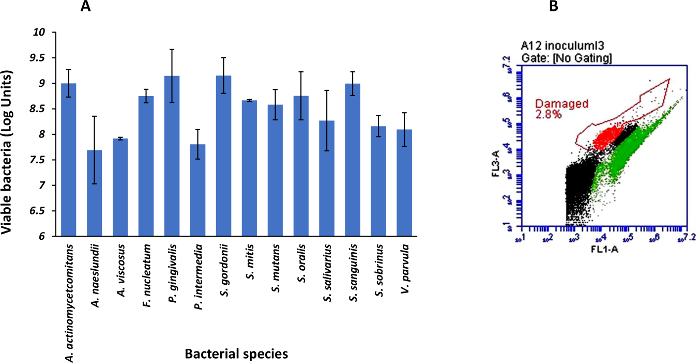
Figure 1: Cell viability of the bacteria composing the multispecies microbial community at the beginning of the in vitro gastrointestinal digestion. (A) Viable cell counts (log units) quantified by live/death staining and flow cytometry (average ± standard deviation, n = 3). (B) Representative flow cytometry plot of the multispecies microbial community after 48 h of co-culture. Dots in green, red, and black represent viable, damaged and background or non-classified bacteria, respectively. Please click here to view a larger version of this figure.
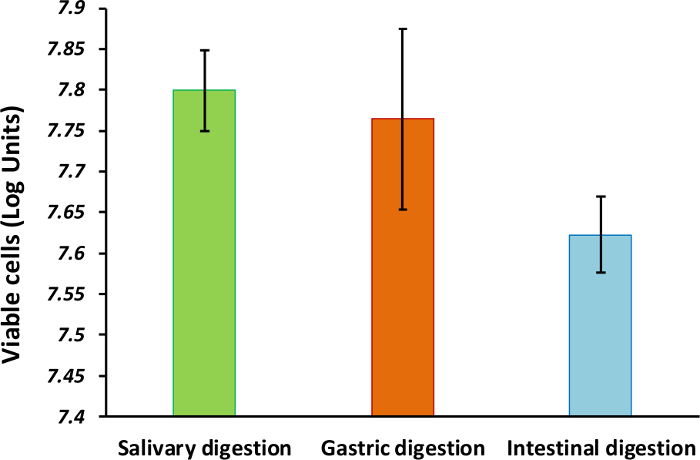
Figure 2: Cell viability of the bacteria after the simulated gastrointestinal digestion. Bars represent the number of viable cells (log units) after the oral, gastric, and small intestinal digestion steps (average ± standard deviation, n = 3). Please click here to view a larger version of this figure.
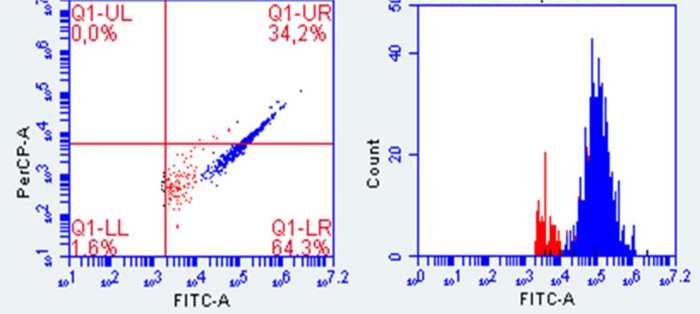
Figure 3: Representative plot of viable bacterial cells in the mucosal interphase. Red dots represent the background and blue dots the viable cells. Flow cytometry conditions were established based on Props et al. (2016)19. Mucosal samples were diluted 1:100 (v/v) and stained with Sybr Green/Propidium Iodide for 15 min, and 100 µL of sample was measured at FL1: 533/30 nm, FL2: 585/40 nm, FL3: >670 nm long pass, and FL4: 675/25 nm. Please click here to view a larger version of this figure.

Figure 4: Colony forming units (CFU) grown from the cellular debris in modified BHI plates. Three biological replicates were used for the bacterial adhesion assay and three technical replicates were performed for the CFU quantification. Two replicate plates containing -1 to -6 dilutions of the cell debris (S7) are shown in the figure. Please click here to view a larger version of this figure.
| Cell culture | Adherent/ Suspension | Culturing Medium | General supplements | Specific supplements | Doubling time | Incubation conditions |
| Caco-2 (ECACC 86010202) | Adherent | Dulbecco’s Modified Eagle Medium (DMEM) with 4.5 g/L glucose | Inactivated fetal bovine serum (10% v/v) Penicillin (100 U/mL)* Streptomycin (0.1 mg/mL)* Amphotericin (0.0025 mg/mL)* |
Glutamax (4 mM) Non essential aminoacids (1% v/v) Pyruvate (1 mM) |
65–72 h | 95% humidity and 10% CO2 CO2 incubator (see Table of Materials) |
| HT29-MTX (ECACC 12040401) | Adherent | HEPES (1 mM) | 20–24 h | |||
| * Antibiotics and antifungals were removed from the cell culture media 2 days before starting the assays. | ||||||
Table 1: Description of the cell lines and media composition for cell culture maintenance.
Discussion
The oral microbiome is a key element in human health as recently reported by several authors20,21. Previous findings suggest that the ingestion of saliva containing large loads of bacteria can influence the microbial ecosystem of the small intestine, which is one of the main sites for immune priming. The combination of a static upper gastrointestinal digestion model with the host interface represented by intestinal epithelial and mucus-secreting cells, served to unravel the impact of the microbial component on the host.
In vitro models are basic tools for research, allowing for conducting mechanistic studies and achieving background knowledge intended to reduce, make more efficient, and better target in vivo studies. For this reason, it is critical to ensure purity, viability, and optimal growth of axenic cultures prior to establishing the synthetic microbial community. We recommend evaluating the viability and composition of bacterial communities before exposure to cell cultures, to avoid bias in the analysis. High number of damaged bacteria may hinder the adhesion and further impact the integrity of the cell model. In addition, the use of a trustable source of cell lines will minimize cell line misidentification, contamination, and poor annotation, enhancing experimental reproducibility22.
Including mucus-producing cells enables resemblance to the small intestine, as mucus and mucins provide the first defence line of the gastrointestinal tract23. In addition, the mucosal layer is suitable for bacterial colonisation, allowing for characterising bilateral transport of bacterial metabolites. Moreover, simulated gastrointestinal conditions increase the adhesion ability of Lactobacillus paracasei strains to Caco-2 and mucin24, supporting the relevance of incorporating digestive processes in our model.
Further improvements in the model can be introduced to incorporate the immune host component, as previously reviewed25. However, current co-culture systems of epithelial and immune cells have not been used for host-microbe interaction applications (e.g., evaluation of food bioactive compounds or probiotics), which might indicate that those systems may lack reproducibility25.
Previous research with Caco-2, HT29-MTX or co-cultures assessed the adhesion of probiotic or pathogenic strains to mammalian cells26,27. Using single stains or associations of few microorganisms may provide a limited overview on the host-microbe interactions, and it is not representative of the complexity of the human gastrointestinal tract. Although the use of natural microbial communities can be more representative of the in vivo scenario, they are difficult to characterise and to study. In contrast, our methodology offers the opportunity of assessing multiple host-microbe interactions, using a complex and representative microcosm. The use of a defined synthetic microbial community allows for the generation of a system with reduced complexity28. Changes in the community can be a result of the microenvironmental conditions in the context of individual experiments. Thus, the microbial component of this model can be easily scaled up or down for deconstructing the impact of environment as a selective force. Finally, sampling accessibility guarantees further characterisation of the functional activities of both human cells and the associated microbial community.
In the past years, new in vitro models based on organoid technology have been developed. Organoids are 3D cell cultures that incorporate some of the key features of the represented primary tissue. Although intestinal organoids are a near-physiological system for studying adult stem cells and tissues, such model also has some limitations. Intestinal organoids have limited use in resembling inflammatory responses to infection or drugs, as they lack immune cells. Additionally, organoids are heterogeneous regarding viability, size, and shape, impeding phenotype screening and rendering standardization complex. Despite the limitation of immortalized cell lines for in vitro modelling of healthy tissues, the use of well-characterized and stable cell lines also offers advantages as repeatability, reproducibility, and low cost. These benefits allow for simultaneous screening of several conditions, but the translational use of the data is controversial. Primary cells may be more representative of in vivo physiology; however, they have high inter-individual variability, are not fully characterized, are heterogeneous, and have a limited time span. These factors are a drawback for including multiple conditions or replicates in one assay.
As combining cell lines with complex microbial communities may represent a challenge, we focused on developing a reproducible and repeatable model with high flexibility and applicability in laboratories with basic cell equipment, until more representative models became available and well characterised. We have evaluated the functionality of diverse bacterial communities using these multicompartment models, for instance, for evaluating probiotic behaviour in the upper gastrointestinal tract and for characterising xenobiotic impact on the gut interface. Thus, we propose this model as a base for further assays with a wide variety of probiotics, drugs, or food compounds, within a relevant and defined in vitro ecosystem.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
The authors gratefully acknowledge financial support from the Flanders Research Foundation to Marta Calatayud Arroyo (FWO postdoctoral fellowship-12N2815N). Emma Hernandez-Sanabria is a postdoctoral fellow supported by Flanders Innovation and Entrepreneurship (Agentschap voor Innovatie door Wetenschap en Technologie, IWT).
Materials
| STRAINS | |||
| Aggregatibacter actinomycetemcomitans | American Type Culture Collection | ATCC 43718 | |
| Fusobacterium nucleatum | American Type Culture Collection | ATCC 10953 | |
| Porphyromonas gingivalis | American Type Culture Collection | ATCC 33277 | |
| Prevotella intermedia | American Type Culture Collection | ATCC 25611 | |
| Streptococcus mutans | American Type Culture Collection | ATCC 25175 | |
| Streptococcus sobrinus | American Type Culture Collection | ATCC 33478 | |
| Actinomyces viscosus | American Type Culture Collection | ATCC 15987 | |
| Streptococcus salivarius TOVE-R | |||
| Streptococcus mitis | American Type Culture Collection | ATCC 49456 | |
| Streptococcus sanguinis | BCCM/LMG Bacteria Collection | LMG 14657 | |
| Veillonella parvula | Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures | DSM 2007 | |
| Streptococcus gordonii | American Type Culture Collection | ATCC 49818 | |
| CELL LINES | |||
| Caco-2 cells | European Collection of Authenticated Cell Cultures | 86010202 | |
| HT29-MTX cells | European Collection of Authenticated Cell Cultures | 12040401 | |
| REAGENTS AND CONSUMABLES | |||
| Brain Heart Infusion (BHI) broth | Oxoid | CM1135 | |
| Blood Agar 2 | Oxoid | CM0055 | Blood Agar medium |
| Menadione | Sigma | M9429 | |
| Hemin | Sigma | H9039 | |
| 5% sterile defibrinated horse blood | E&O Laboratories Ltd, | P030 | |
| InnuPREP PCRpure Kit | Analytik Jena | 845-KS-5010250 | PCR purification kit |
| Big Dye | Applied Biosystems | 4337454 | Dye for sequencing |
| ABI Prism BigDye Terminator v3.1 cycle sequencing kit | Applied Biosystems | 4337456 | |
| SYBR Green I | Invitrogen | S7585 | |
| Propidium Iodide | Invitrogen | P1304MP | |
| T25 culture flasks uncoated, cell-culture treated, vented, sterile | VWR | 734-2311 | |
| Trypsin-EDTA solution | Sigma-Aldrich | T3924-100ML | |
| Trypan Blue solution 0.4%, liquid, sterile-filtered |
Sigma-Aldrich | T8154 | |
| PBS | Gibco | 14190250 | |
| DMEM cell culture media, with GlutaMAX and Pyruvate | Life technologies | 31966-047 | |
| Corning Transwell polyester membrane cell culture inserts | Sigma-Aldrich | CLS3450-24EA | |
| Mucin from porcine stomach Type II | Sigma-Aldrich | M2378 | |
| Inactivated fetal bovine serum | Greiner Bio One | 758093 | |
| Antibiotic-Antimycotic (100X) | Gibco | 15240062 | |
| Triton X 100 for molecular biology | Sigma-Aldrich | T8787 | |
| DPBS without calcium, magnesium | Gibco | 14190-250 | |
| Pierce LDH Cytotoxicity Assay Kit | Thermo Fisher Scientific | 88953 | |
| Corning HTS Transwell-24 well, pore size 0.4 µm | Corning Costar Corp | 3450 | |
| Nuclease-free water | Serva Electrophoresis | 28539010 | |
| EQUIPMENT | |||
| Neubauer counting chamber improved | Carl Roth | T729.1 | |
| BD Accuri C6 Flow cytometer | BD Biosciences | 653118 | |
| PowerLyzer 24 Homogenizer | MoBio | 13155 | |
| T100 Thermal Cycler | BioRad | 186-1096 | |
| Flush system | Custom made | – | |
| InnOva 4080 Incubator Shaker | New Brunswick Scientific | 8261-30-1007 | Shaker for 2.10 |
| Memmert CO2 incubator | Memmert GmbH & Co. | ICO150med | |
| Millicell ERS (Electrical Resistance System) | EMD Millipore, Merck KGaA | MERS00002 | |
| Millipore Milli-Q academic, ultra pure water system | Millipore, Merck KGaA | – | |
| Shaker (ROCKER 3D basic) | IKA | 4000000 | Shaker for 6.10 |
Riferimenti
- Sender, R., Fuchs, S., Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biology. 14, e1002533 (2016).
- Kelly, D., King, T., Aminov, R. Importance of microbial colonization of the gut in early life to the development of immunity. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. , 58-69 (2007).
- Aas, J. A., Paster, B. J., Stokes, L. N., Olsen, I., Dewhirst, F. E. Defining the normal bacterial flora of the oral cavity. Journal of Clinical Microbiology. 43, 5721-5732 (2005).
- Marsh, P. D., Head, D. A., Devine, D. A. Dental plaque as a biofilm and a microbial community-Implications for treatment. Journal of Oral Biosciences. 57, 185-191 (2015).
- Socransky, S. S., Haffajee, A. D., Cugini, M. A., Smith, C., Kent, R. L. Microbial complexes in subgingival plaque. Journal of Clinical Periodontology. 25, 134-144 (1998).
- Haffajee, A. D., et al. Subgingival microbiota in healthy, well-maintained elder and periodontitis subjects. Journal of Clinical Periodontology. 25, 346-353 (1998).
- Venema, K. Microbial metabolites produced by the colonic microbiota as drivers for immunomodulation in the host. The FASEB Journal. 27, (2013).
- Marzorati, M., et al. The HMI (TM) module: A new tool to study the Host-Microbiota Interaction in the human gastrointestinal tract in vitro. BMC Microbiology. 14, 133 (2014).
- Tanzer, J. M., Kurasz, A. B., Clive, J. Competitive displacement of mutans streptococci and inhibition of tooth-decay by Streptococcus-Salivarius Tove-R. Infection and Immunity. 48, 44-50 (1985).
- Slomka, V., et al. Nutritional stimulation of commensal oral bacteria suppresses pathogens: the prebiotic concept. Journal of Clinical Periodontology. 44, 344-352 (2017).
- Hernandez-Sanabria, E., et al. In vitro increased respiratory activity of selected oral bacteria may explain competitive and collaborative interactions in the oral microbiome. Frontiers in Cellular and Infection Microbiology. 7, 235 (2017).
- Alvarez, G., Gonzalez, M., Isabal, S., Blanc, V., Leon, R. Method to quantify live and dead cells in multi-species oral biofilm by real-time PCR with propidium monoazide. AMB Express. 3, (2013).
- Ehsani, E., et al. Initial evenness determines diversity and cell density dynamics in synthetic microbial ecosystems. Scientific Reports. 8, 340 (2018).
- Hernandez-Sanabria, E., et al. Correlation of particular bacterial PCR-denaturing gradient gel electrophoresis patterns with bovine ruminal fermentation parameters and feed efficiency traits. Applied and Environmental Microbiology. 76, 6338-6350 (2010).
- Masters, J. R., Stacey, G. N. Changing medium and passaging cell lines. Nature Protocols. 2, 2276-2284 (2007).
- Calatayud, M., Velez, D., Devesa, V. Metabolism of inorganic arsenic in intestinal epithelial cell lines. Chemical Research in Toxicology. 25, 2402-2411 (2012).
- Minekus, M., et al. A standardised static in vitro digestion method suitable for food – an international consensus. Food & Function. 5, 1113-1124 (2014).
- Berney, M., Hammes, F., Bosshard, F., Weilenmann, H. U., Egli, T. Assessment and interpretation of bacterial viability by using the LIVE/DEAD BacLight kit in combination with flow cytometry. Applied and Environmental Microbiology. 73, 3283-3290 (2007).
- Props, R., Monsieurs, P., Mysara, M., Clement, L., Boon, N. Measuring the biodiversity of microbial communities by flow cytometry. Methods in Ecology and Evolution. 7, 1376-1385 (2016).
- Yamashita, Y., Takeshita, T. The oral microbiome and human health. Journal of Oral Science. 59, 201-206 (2017).
- Wade, W. G. The oral microbiome in health and disease. Pharmacological Research. 69, 137-143 (2013).
- Yu, M., et al. A resource for cell line authentication, annotation and quality control. Nature. 520, 307 (2015).
- Pelaseyed, T., et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunological Reviews. 260, 8-20 (2014).
- Bengoa, A. A., et al. Simulated gastrointestinal conditions increase adhesion ability of Lactobacillus paracasei strains isolated from kefir to Caco-2 cells and mucin. Food Research International. 103, 462-467 (2018).
- Kleiveland, C. R., et al., Verhoeckx, K., et al. . The Impact of Food Bioactives on Health: in vitro and ex vivo models. , 197-205 (2015).
- Laparra, J. M., Sanz, Y. Comparison of in vitro models to study bacterial adhesion to the intestinal epithelium. Letters in Applied Microbiology. 49, 695-701 (2009).
- Gagnon, M., Berner, A. Z., Chervet, N., Chassard, C., Lacroix, C. Comparison of the Caco-2, HT-29 and the mucus-secreting HT29-MTX intestinal cell models to investigate Salmonella adhesion and invasion. Journal of Microbiological Methods. 94, 274-279 (2013).
- Großkopf, T., Soyer, O. S. Synthetic microbial communities. Current Opinion in Microbiology. 18, 72-77 (2014).
