Correlativos luz microscopia electrónica (CLEM) para el seguimiento y proyección de imagen proteína Viral asociada a estructuras en las células inmovilizadas de Cryo
Summary
Un método de microscopía electrónica correlativos (CLEM) se aplica a estructuras intracelulares inducida por virus de imagen mediante microscopia electrónica (EM) en las células que son previamente seleccionadas por microscopía de luz (LM). EM y LM se combinan como un enfoque de imagen híbrida para lograr una visión integrada de las interacciones virus-huésped.
Abstract
Debido a su alta resolución, la microscopia electrónica (EM) es una herramienta indispensable para los virólogos. Sin embargo, una de las principales dificultades al analizar infectado o transfected las células por medio de EM son la baja eficiencia de la infección o transfección, lo que dificulta el examen de estas células. Para superar esta dificultad, microscopía de luz (LM) se puede realizar primero para asignar la subpoblación de células infectadas o transfected. Así, aprovechando el uso de proteínas fluorescentes (FPs) unida a las proteínas virales, LM se utiliza aquí para registrar las posiciones de las células “positivo-transfected”, expresando una FP y creciendo sobre un soporte con un patrón alfanumérico. Posteriormente, las células son procesadas más de EM con alta presión (HPF) de congelación, congelación de resina incrustación y sustitución (FS). El paso de congelación ultra rápido garantiza la preservación excelente de la membrana de las celdas seleccionadas que luego pueden ser analizadas a nivel ultraestructural por microscopia electrónica de transmisión (TEM). Aquí, se proporciona un flujo de trabajo de microscopía de electrones luz correlativa paso a paso (CLEM), describiendo la preparación de la muestra, la proyección de imagen y correlación en el detalle. El diseño experimental se puede aplicar también para hacer frente a muchas preguntas de Biología de la célula.
Introduction
La idea de combinar dos modalidades de microscopía para obtener una mejor imagen de un proceso biológico específico es bastante vieja. Así, el primer estudio acerca de los virus con “microscopía correlativa” se publicó en 1960 como dos publicaciones separadas1,2. En ese estudio, los autores analizaron los cambios en la morfología del núcleo inducida por adenovirus por medio de dos técnicas de microscopía. En la primera publicación, observaciones de microscopía electrónica (EM) que describe los detalles morfológicos asociados con la infección del adenovirus fueron reportados1. En una segunda publicación, las diferentes estructuras observadas por EM se correlacionaron con las imágenes de microscopía de luz (LM) de patrones de tinción histoquímicos, para definir la naturaleza de las estructuras previamente observado por EM2.
En estos estudios iniciales, sin embargo, sus observaciones se realizaron utilizando diferentes células infectadas como experimentos independientes. La “correlación”, de hecho, iba como la combinación de información proveniente de dos modalidades de imágenes para comprender un fenómeno, comparando todos los hallazgos que han sido obtenidos con diferentes ensayos para entender una determinada biológica proceso.
En la actualidad, la microscopía correlativa del término, también conocido como correlativa luz y la microscopia (CLEM), se aplica a un número creciente de métodos (revisado en referencias3,4,5), con la concordancia que ambas técnicas de imagen (EM y LM) se llevan a cabo en la misma muestra. Resultados de la combinación de ambos métodos, de tal modo, en un análisis multimodal, multiescalar y multidimensional de la muestra3. Las ventajas son que LM puede proporcionar una amplia visión de muchas células diferentes, lo que permite la identificación de subpoblaciones de células expresan una proteína o proteínas de interés dentro de una población celular heterogénea. EM supera el límite de resolución de LM, proporcionando una mayor resolución de imagen de un evento en particular intracelular. Además, la EM permite la visualización del contexto subcelular no fluorescente, incluyendo todos los organelos de membrana enlazado, grandes complejos macromoleculares (por ejemplo, los ribosomas, centriolos, etc) y elementos citoesqueléticos, así proporcionando información espacial adicional, el llamado “espacio de referencia”6y dar contexto a la mancha fluorescente detectada por LM.
Durante los últimos años, CLEM se ha convertido en una herramienta poderosa no sólo para biólogos de la célula5, sino también para los virólogos (revisados en la referencia7) dispuestos a comprender la interacción virus-célula compleja que conducen a una propagación de virus exitoso. Por lo tanto, entender cómo los virus modifican las membranas celulares y orgánulos para su propio beneficio es esencial para el desarrollo de fármacos antivirales para erradicar virus patógenos.
Aquí, CLEM describe un método es que permite la detección de LM de las células que expresan proteínas virales que unida a una proteína fluorescente (FP). Estas células son posteriormente cryo-inmovilizado y más preparados para análisis ultraestructural por microscopia electrónica de transmisión (TEM) para obtener nuevos conocimientos sobre cómo la expresión de estas proteínas reorganizar las membranas intracelulares (figura 1). CLEM ha llevado a cabo con células químicamente fijas en la mayoría de los estudios de virología publicados a fecha8,9,10,11,12,13,14 ,15,16,17,18,19. Esto es principalmente debido a la necesidad de inactivar el material infeccioso por razones de bioseguridad en nivel 2 de bioseguridad y laboratorios (BSL-2 y BSL-3)-3, donde crio-inmovilización de células no es generalmente posible. Para aquellas preguntas que requieren una conservación óptima de las membranas celulares, vitrificación mediante alta presión (HPF) de congelación, sin embargo, recomienda20. En estos casos, se puede aplicar el protocolo de CLEM descrito aquí. Interesante, especialmente cuando se trabaja con muestras infecciosas, HPF puede realizarse en muestras que han sido previamente inactivadas químicamente, por ejemplo en laboratorios BSL-2 y BSL-3. La combinación de fijación química seguida de HPF es una posibilidad de por lo menos parcialmente se benefician de las ventajas de la criopreservación métodos21,22.
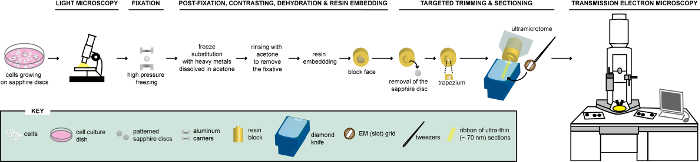
Figura 1 : Representación esquemática del flujo de trabajo para el análisis de las células a través de CLEM. Las células crecen en discos zafiro estampados primero son analizadas por LM para localizar las células expresaban FPs antes de su tratamiento para la EM. Una vez localizado, las células se fijan inmediatamente por HPF y FS para posteriormente ser embebido en resina. En la polimerización de la resina, el soporte donde fueron creciendo las células (= discos zafiro) debe extraerse el bloque de resina. El bloque que contiene las células incrustadas se recorta a un pequeño trapecio que las células restantes, expresando FPs, se seccionaron con un cuchillo de diamante. Secciones ultrafinas son recogidas en rejillas de ranura y más examinadas por TEM para obtención de datos ultraestructurales de estas células. Esta cifra es adaptada y modificada con permiso de referencia29. Haga clic aquí para ver una versión más grande de esta figura.
Protocol
Representative Results
Discussion
La metodología de CLEM presentada aquí para estudiar el impacto de una expresión de la proteína viral en las membranas celulares se ha utilizado con éxito antes de dilucidar el VHC asociados de replicación de las estructuras, principalmente21de la vesículas (DMV) de doble membrana, así como determinar los bloques de edificio crítico requeridos para formar estas estructuras HCV-inducido23. Tenga en cuenta que en nuestro primer trabajo con CLEM para el estudio de la replicación del VHC21, se aplicó una versión ligeramente modificada del protocolo descrito aquí. En ese estudio, discos zafiro grueso convencional 0.05 mm, sin patrón alfanumérico, se utiliza en el que un patrón de referencia fue creado por carbón capa con una cuadrícula de buscador en la parte superior (véase Tabla de materiales). Esta versión del actual protocolo puede eventualmente aplicar, con la ventaja que el “Un” portador de HPF puede utilizarse directamente, sin necesidad de cortar hacia abajo como en la figura 3A. Por otra parte, como se menciona en el protocolo, un más grueso “A” portador puede ser utilizado (véase Tabla de materiales) con discos zafiro estampados, sin la necesidad de un operador “B”.
Curiosamente, este protocolo se puede aplicar no sólo para estudiar muestras de BSL-1, tales como las células transfected con proteínas virales según lo descrito aquí y en otros lugares19,23, sino también para el estudio de células infectadas por virus. Aunque trabajo con patógenos humanos está generalmente restringido a laboratorios BSL-2 y BSL-3, en algunos países es posible realizar cryo-inmovilización en estas condiciones de bioseguridad. En los BSL-2 y BSL-3 laboratorios donde no es posible, debido a las regulaciones locales o a la ausencia de una máquina de HPF vitrificación, células infectadas por virus pueden todavía prepararse utilizando este método si se realiza fijación química con aldehídos por adelantado, es decir, antes para dejar las instalaciones BSL-2 o BSL-3. Además, aldehídos que se apagará inmediatamente después de la fijación para evitar la fluorescencia, mientras que el resto del protocolo es idéntico al descrito aquí. Esta técnica puede considerarse redundante porque las células se fijan dos veces y químicamente mediante vitrificación. Sin embargo, este protocolo de fijación doble de hecho conduce a una mucho mejor preservación de la DMV HCV-inducido en comparación con el DMV en células sometidas a fijación química solo21.
Para más modificaciones y resolución de problemas el lector es referidos a las notas a lo largo de la parte de protocolo de este manuscrito. Estas notas describen errores a evitar, así como las alternativas para superar posibles dificultades que pueden surgir al realizar este método.
El principal requisito para la aplicación de esta técnica es una máquina de HPF. Cuando crio-inmovilizar las células vía HPF no es factible (debido a la ausencia de una máquina de HPF) o no es necesario (cuando la preservación de la membrana no es necesario ser óptimos), las células pueden ser químicamente fijo y posteriormente preparado y analizadas por EM8, 9,10,11,12,13,14,15,16,17,18 ,19. Esta opción no requiere el uso de discos zafiro, pero platos de cultivo celular con patrones cuadriculados para la reubicación de las células o grupos de células. La principal ventaja del uso de estos platos es su diámetro más grande en comparación con discos zafiro, lo que permite la proyección de las superficies más grandes. Por lo tanto, la aplicación de este protocolo de CLEM ha sido aplicada con éxito para estudiar el efecto de un compuesto antiviral contra VHC15 o para visualizar los cambios de la membrana inducidos por las proteínas no estructurales de los norovirus19. Otra cuestión que puede limitar los resultados de este método es la ausencia de un dispositivo comercial de FS. En este caso, sistemas de FS caseros básicos pueden ser utilizados en su lugar. Aunque los dispositivos automáticos de FS podrían reducir manejo de contratiempos, dispositivos caseros se utilizan con éxito por ejemplo en laboratorios de Paul Walther y30 de Kent McDonald .
Con respecto a la fijación química, el protocolo descrito aquí asegura una conservación óptima de las estructuras intracelulares20. Por lo tanto, en el caso de los dispositivos mencionados de HPF y FS están disponibles, vitrificado de las células de interés sería preferido.
Alternativas futuras a este enfoque de CLEM incluyen la posibilidad de utilizar este método no sólo para adquirir información 2D a nivel ultraestructural, sino también para obtener información 3D sobre la arquitectura de las membranas y organelas alteraciones causadas por virus. Los métodos EM 3D, incluyendo tomografía de electrón (ET) y ion enfocado viga-microscopía electrónica (SEM-FIB) (ampliamente descrito en29), podrían aplicarse también a las células que han sido preparadas siguiendo este protocolo actual21, 31. Además, 3D podría también obtenerse información en el nivel de LM, cuando se utiliza un microscopio confocal, que permite la adquisición de pilas de z. De hecho, esta opción se recomienda cuando una correlación exacta entre conjuntos de datos EM y LM es deseada (véase por ejemplo17). La información incluida en 3D z-pilas ayuda a mejorar la correlación con las imágenes 2D de TEM. Así, en tal escenario, el mejor montaje de imágenes EM y LM puede ser seleccionado y después sometido a uno de los software de correlación disponible, por ejemplo el plugin de CE-CLEM de ICY (http://icy.bioimageanalysis.org/)32 o el plugin de las correspondencias de la señal de Imagen J (http://imagej.net/Landmark_Correspondences), resultando en la generación de imágenes de LM-EM se superponen.
Cuando la información temporal es necesario para comprender la cinética de un determinado evento, proyección de imagen de Time-lapse puede utilizarse para monitorear la dinámica de las células vivas en combinación con EM. Durante el evento de interés, las células se fijan inmediatamente, generando una “instantánea congelada” que puede ser posteriormente analizada por EM, proporcionando información ultraestructural detallada sobre ese momento en el tiempo de inmovilización. Para obtener esa “instantánea inmovilizada”, después de la observación en tiempo real, las células pueden ser químicamente fijo33 o inmovilizada de cryo6. Ya que muchos procesos celulares ocurren más rápidamente que los procesos de difusión de fijación química, si es posible, debe realizarse congelación ultra rápida. Sin embargo, es importante tener en cuenta que las máquinas de HPF difieren en su tiempo efectivo resolución34.
Además, aunque este protocolo ha sido diseñado para la fijación de las células en una resina de epoxy, las células pueden ser también incrustadas en las resinas de baja viscosidad, tales como Lowicryls, White LR o LR oro. El uso de estos medio de inclusión permite preservar la antigenicidad35,36, así como la fluorescencia37,38 y, por lo tanto, se utilizan sobre todo para incrustar la inmunomarcación en sección39 , 40 y en la sección CLEM41,42,43,44, donde el LM se realiza después de la incrustación. Ambos enfoques (inmuno-EM y en la sección CLEM) deben ser cruciales para aquellos experimentos en que las estructuras de la característica no se pueden encontrar fácilmente mediante TEM o como control contra miscorrelation entre LM y EM señales. Así mismo, etiquetado con anticuerpos que pueden ser visualizados por ambas modalidades de imágenes (LM y EM) puede llevarse a cabo45 para, por ejemplo, identificar células transfected en LM (la pre-inserción etapa) y alcanzar una localización mucho más exacta de la Señal GFP a través de su etiqueta específica por inmuno-EM (etapa posterior incrustación). Debe tenerse en cuenta, sin embargo, esa permeabilización se lleva a cabo antes de LM para permitir el acceso de los anticuerpos al espacio intracelular, que puede resultar en una conservación óptima de la arquitectura de la célula en el nivel de EM. Curiosamente, este protocolo está también bien adaptado para experimentos de varios colores que se logra con el uso de otras etiquetas fluorescentes, que no sean de GFP (como se muestra aquí). En conclusión, existen muchas supuestas posibilidades de adaptación de este protocolo, tanto en el LM o en los lados de la EM, según las preguntas que se están abordando. Para una completa descripción de otros protocolos alternativos el lector se refiere a5,46. Independientemente de cómo se combinan las modalidades de microscopía, juntos el resultado es un aumento de la información, lo que nos permite comprender mejor cómo el virus y sus proteínas interactúan con sus anfitriones en la vida real.
El paso más crítico dentro de este método es la colección de secciones seriadas de las células de interés. Como se destacó en la sección de resultados representativos, esto requiere personal experto, así como mucha paciencia. Lo importante, este paso es fundamental para encontrar las células en el nivel de EM por dos razones. En primer lugar, en este tipo de protocolo de CLEM utilizando la pre-inserción LM, las coordenadas sólo son visibles en el plano de la LM y en la cara del bloque de resina después de la incrustación. Sin embargo, no serán visibles en las secciones por TEM. Por lo tanto, objetivo recortar en el bloque hacia abajo a las regiones de interés (ROIs) debe realizarse cuidadosamente con una cuchilla de afeitar para garantizar que las secciones que se obtienen posteriormente contienen las células que expresan un determinado PF. En segundo lugar, varias secciones de la “exploración” es necesaria encontrar el mejor recubrimiento entre el LM y las adquisiciones de EM. En contraste con métodos que LM se realiza en la etapa de la inclusión, en este caso el recubrimiento LM-EM no es tan preciso. La precisión baja superposición es debido a diferencias en la resolución axial entre LM y EM, contracción durante el procesamiento de la muestra de EM y la compresión durante el seccionamiento42. Sin embargo, métodos de seguimiento eficiente, como el uso de lugares de interés, ayudan a encontrar las células detrás. Esto incluye la posición de una celda a otra, así como la forma de las células y su núcleo. En este sentido, como se explica en el protocolo, DIC imágenes proporcionan información “anatómica” de las células que son fundamentales para mejorar la correlación. Alternativamente, los núcleos u otros orgánulos celulares así reconocible (como las mitocondrias o lípido gotitas) puede ser teñido antes de LM y utilizado como puntos de referencia.
Por último, cabe mencionar que aunque este manuscrito se centra en el uso de esta técnica para estudios de virología, puede ampliarse el alcance de este diseño experimental para abordar cuestiones biológicas más generales.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
Estamos muy agradecidos a los miembros del personal de la microscopia electrónica base instalaciones (EMCF) en el EMBL (Heidelberg) y en la Universidad de Heidelberg. También quisiéramos agradecer a Ulrike Herian, Stephanie Kallis y Andrea Hellwig (Universidad de Heidelberg), Eberhardt Schmidt y Renate Kunz (Universidad de Ulm) de asistencia técnica de expertos. Trabajo por R.B. y su equipo (H.U. y B. I.R.) fue apoyada por la Deutsche Forschungsgemeinschaft, SFB1129, TP11 y TRR83, TP13.
Materials
| UV Crosslinker | Stratagene | 400072 | Stratalinker 1800, 230Vac, equipped with 254-nm UV light bulbs, 8-watts/each. |
| Patterned sapphire discs | Engineering Office M. Wohlwend, Sennwald, Switzerland | 627 | They have a 0.3-mm diameter and are 0.16-mm thick, as well as an an etched alphanumeric pattern to allow for re-location of the cells. They can be only ordered per e-mail: martin-wohlwend@bluewin.ch. |
| Slim and long tweezers | Electron Microscopy Sciences, Hatfield, PA, USA | 72919-SS | "Style SS", for handling sapphire discs. |
| Cell culture medium | Thermo Fisher Scientific, Waltham, MA, USA | 41965-062 | Dulbecco's modified Eagle medium (DMEM) supplemented with 2 mM L-glutamine, nonessential amino acids, 100 units penicillin per ml, 100 µg streptomycin per ml and 10% fetal calf serum (DMEM complete, see below). Package containing 10 bottles, 500 ml/each. |
| L-glutamine | Thermo Fisher Scientific, Waltham, MA, USA | 25030-123 | Package containing 20 bottles of 100 ml/each. |
| Nonessential amino acids | Thermo Fisher Scientific, Waltham, MA, USA | 11140-068 | Package containing 20 bottles of 100 ml/each. |
| Penicillin/Streptomycin | Thermo Fisher Scientific, Waltham, MA, USA | 15140-163 | Package containing 20 bottles of 100 ml/each. |
| Fetal calf serum | Sigma Aldrich, St. Louis, MI, USA | F7524 | Bottle of 50 ml. |
| Inverted microscope | Olympus Deutschland, Hamburg, Germany | CKX41 | It is an inverted microscope with trinocular head options and fluorescence upgrade capability. |
| Huh7-Lunet cells stably expressing the T7 RNA-polymerase | Avaliable at Prof. Dr. Ralf Bartenschlager laboratory | Not available | Contact Prof. Bartenschlager at: ralf.bartenschlager@med.uni-heidelberg.de. |
| Mirus TransIT-LT1 Transfection Reagent | Mirus Bio LLC, Madison, USA | MIR 2304 | Broad spectrum, low toxicity, DNA transfection reagent. Vial of 0.4 ml. |
| Inverted widefield fluorescence microscope | Carl Zeiss Microscopy GmbH, Germany | Zeiss Observer.Z1 | Inverted fluorescence microscope for experiments involving living cell cultures. |
| 1-hexadecene | Merck, Darmstadt, Germany | 8220640100 | Bottle of 100 ml. |
| "A" aluminium holders for high pressure frezing (HPF) | Engineering Office M. Wohlwend, Sennwald, Switzerland | 241 | This is the holder that has been used for this protocol and has 2 different depths of 0.1 and 0.2-mm. It can be only ordered per e-mail: martin-wohlwend@bluewin.ch. |
| "B" aluminium holders for HPF | Engineering Office M. Wohlwend, Sennwald, Switzerland | 242 | This is the holder that has been used for this protocol.It is 0.3-mm thick.They can be only ordered per e-mail: martin-wohlwend@bluewin.ch. |
| "A" aluminium holder for HPF (special for working with patterned sapphire discs) | Engineering Office M. Wohlwend, Sennwald, Switzerland | 737 | This is the holder than can be used alone with the patterned sapphire disc (as an alternative to the current protocol), without the need of a "B" holder or the edition of the "A" holder. It is 0.84-mm thick. It can be only ordered per e-mail: martin-wohlwend@bluewin.ch. |
| Sapphire discs | Engineering Office M. Wohlwend, Sennwald, Switzerland | 405 | These sapphires discs are the "conventional" one, with a 0.3-mm diameter and a thickness of 0.05 mm . They can be also used for CLEM as an alternative instead of patterned sapphire discs. |
| Finder grids | Electron Microscopy Sciences, Hatfield, Philadelphia, USA | LF135-Cu | These 135 mesh grids are used to create an alphanumeric pattern on "conventional" sapphire discs (described above) via carbon coating, so that they can be used for CLEM. 100 grids/vial. |
| HPF machine | ABRA Fluid AG, Widnau, Switzerland | HPM 01 | This HPF machine has been used for this protocol. |
| HPF machine | Leica Microsystems, Vienna, Austria | EMPACT 2 | "EMPACT 2", as an alternative to the use of the HPM machine that it has been used in this protocol (described above). |
| Cryo-tubes | Thermo Fisher Scientific, Waltham, MA, USA | Z763667-500EA | For long term-storage of cryo-immobilized samples in liquid nitrogen. Package containing 500 cryovials. |
| Liquid nitrogen dewar | Cole-Parmer GmbH, Wertheim, Germany | GZ-05094-60 | For long term-storage of cryo-immobilized samples in liquid nitrogen, equipped with cryo-racks with a capacity of 1600 cryo-tubes. |
| Automatic freeze substitution (AFS) machine | Leica Microsystems, Vienna, Austria | EM AFS2 | This machine performs freeze substitution and progressive lowering of temperature (PLT) techniques and allows low temperature embedding and polymerization of resins. |
| Osmium tetroxide (OsO4) | Electron Microscopy Sciences, Hatfield, PA, USA | 19150 | 4% aqueous solution, one box containing 10 x 2 ml ampules. |
| Uranyl-Acetate (UA) | Serva, Heidelberg, Germany | 77879.01 | Bottle containing 25 grs of (UA)-2 H2O. |
| Sonicator | Bandelin Electronic, Berlin, Germany | 329 | "Sonorex Super RK 31" is a high-power ultrasonic cleaning bath for aqueous cleaning solution that is used in this protocol to mix OsO4 and Ua when preparing the FS medium. |
| Glass-distilled acetone | Electron Microscopy Sciences, Hatfield, PA, USA | 10015 | Bottle of 100 ml. |
| Stereomicroscope | Leica Microsystems, Vienna, Austria | Leica M80 | Routine stereomicroscope for daily inspections, equipped with a camera for capturing images. |
| Epoxy resin | Electron Microscopy Sciences, Hatfield, PA, USA | 14120 | Embed 812 is a kit containing several components that must be mix together in the proportions given by the manufacturers. |
| Flow through rings | Leica Microsystems, Vienna, Austria | 16707157 | Package containing 100 pieces. |
| Reagent baths | Leica Microsystems, Vienna, Austria | 16707154 | Package containing 100 pieces. |
| Ultramicrotome | Leica Microsystems, Vienna, Austria | Leica EM UC7 | Ultramicrotome for preparation of semi- and ultrathin sections at room temperature. |
| Ultra 35° diamond knife | Diatome Ltd., Nidau Switzerland |
AGG339-735 | This knife have an edge length of 3 mm. |
| Ultra fine tweezers | Electron Microscopy Sciences, Hatfield, PA, USA | E78318 | "Style 3X", for handling EM grids. |
| EM slot grids | Electron Microscopy Sciences, Hatfield, PA, USA | G2010-Cu | 100 grids/vial. |
| EM grid storage box | Electron Microscopy Sciences, Hatfield, PA, USA | 71155 | It has capacity for 100 grids. |
Riferimenti
- Morgan, C., Godman, G. C., Breitenfeld, P. M., Rose, H. M. A correlative study by electron and light microscopy of the development of type 5 adenovirus. I. Electron microscopy. Journal of Experimental Medicine. 112, 373-382 (1960).
- Godman, G. C., Morgan, C., Breitenfeld, P. M., Rose, H. M. A correlative study by electron and light microscopy of the development of type 5 adenovirus. II. Light microscopy. Journal of Experimental Medicine. 112, 383-402 (1960).
- Caplan, J., Niethammer, M., Taylor, R. M., Czymmek, K. J. The power of correlative microscopy: multi-modal, multi-scale, multi-dimensional. Current Opinion in Structural Biology. 21, 686-693 (2011).
- de Boer, P., Hoogenboom, J. P., Giepmans, B. N. Correlated light and electron microscopy: ultrastructure lights up. Nature Methods. 12, 503-513 (2015).
- Müller-Reichert, T., Verkade, P. . Correlative light and electron microscopy III, First edition. , (2017).
- Brown, E., Mantell, J., Carter, D., Tilly, G., Verkade, P. Studying intracellular transport using high-pressure freezing and Correlative Light Electron Microscopy. Seminars in Cell and Developmental Biology. 20, 910-919 (2009).
- Bykov, Y. S., Cortese, M., Briggs, J. A., Bartenschlager, R. Correlative light and electron microscopy methods for the study of virus-cell interactions. FEBS Letters. , (2016).
- Spuul, P., et al. Assembly of alphavirus replication complexes from RNA and protein components in a novel trans-replication system in mammalian cells. Journal of Virology. 85, 4739-4751 (2011).
- Nagel, C. H., Dohner, K., Binz, A., Bauerfeind, R., Sodeik, B. Improper tagging of the non-essential small capsid protein VP26 impairs nuclear capsid egress of herpes simplex virus. PLoS One. 7, 44177 (2012).
- Sharma, M., Kamil, J. P., Coughlin, M., Reim, N. I., Coen, D. M. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. Journal of Virology. 88, 249-262 (2014).
- Kallio, K., et al. Template RNA length determines the size of replication complex spherules for Semliki Forest virus. Journal of Virology. 87, 9125-9134 (2013).
- Martinez, M. G., Snapp, E. L., Perumal, G. S., Macaluso, F. P., Kielian, M. Imaging the alphavirus exit pathway. Journal of Virology. 88, 6922-6933 (2014).
- Lebrun, M., et al. Varicella-zoster virus induces the formation of dynamic nuclear capsid aggregates. Virology. 454-455, 311-327 (2014).
- Madela, K., et al. A simple procedure to analyze positions of interest in infectious cell cultures by correlative light and electron microscopy. Methods in Cell Biology. 124, 93-110 (2014).
- Berger, C., et al. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology. 147, 1094-1105 (2014).
- van der Schaar, H. M., et al. Illuminating the Sites of Enterovirus Replication in Living Cells by Using a Split-GFP-Tagged Viral Protein. mSphere. 1, (2016).
- Vale-Costa, S., et al. Influenza A virus ribonucleoproteins modulate host recycling by competing with Rab11 effectors. Journal of Cell Science. 129, 1697-1710 (2016).
- Wang, L., et al. Visualization of HIV T Cell Virological Synapses and Virus-Containing Compartments by Three-Dimensional Correlative Light and Electron Microscopy. Journal of Virology. 91, (2017).
- Doerflinger, S. Y., et al. Membrane alterations induced by nonstructural proteins of human norovirus. PLOS Pathogens. 13, 1006705 (2017).
- Dahl, R., Staehelin, L. A. High-pressure freezing for the preservation of biological structure: theory and practice. Journal of Electron Microscopy Technique. 13, 165-174 (1989).
- Romero-Brey, I., et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLOS Pathogens. 8, 1003056 (2012).
- Sosinsky, G. E., et al. The combination of chemical fixation procedures with high pressure freezing and freeze substitution preserves highly labile tissue ultrastructure for electron tomography applications. Journal of Structural Biology. 161, 359-371 (2008).
- Romero-Brey, I., et al. NS5A Domain 1 and Polyprotein Cleavage Kinetics Are Critical for Induction of Double-Membrane Vesicles Associated with Hepatitis C Virus Replication. MBio. 6, 00759 (2015).
- Dixit, R., Cyr, R. Cell damage and reactive oxygen species production induced by fluorescence microscopy: effect on mitosis and guidelines for non-invasive fluorescence microscopy. The Plant Journal. 36, 280-290 (2003).
- Jou, M. J., Jou, S. B., Guo, M. J., Wu, H. Y., Peng, T. I. Mitochondrial reactive oxygen species generation and calcium increase induced by visible light in astrocytes. Annals of the New York Academy of Sciences. 1011, 45-56 (2004).
- McDonald, K. L., Morphew, M., Verkade, P., Muller-Reichert, T. Recent advances in high-pressure freezing: equipment- and specimen-loading methods. Methods in Molecular Biology. 369, 143-173 (2007).
- Walther, P., Ziegler, A. Freeze substitution of high-pressure frozen samples: the visibility of biological membranes is improved when the substitution medium contains water. Journal of Microscopy. 208, 3-10 (2002).
- White, J. G., Southgate, E., Thomson, J. N., Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philosophical Transactions of the Royal Society B: Biological Sciences. 314, 1-340 (1986).
- Romero-Brey, I., Bartenschlager, R. Viral Infection at High Magnification: 3D Electron Microscopy Methods to Analyze the Architecture of Infected Cells. Viruses. 7, 6316-6345 (2015).
- McDonald, K. L., Webb, R. I. Freeze substitution in 3 hours or less. Journal of Microscopy. 243, 227-233 (2011).
- Villinger, C., Neusser, G., Kranz, C., Walther, P., Mertens, T. 3D Analysis of HCMV Induced-Nuclear Membrane Structures by FIB/SEM Tomography: Insight into an Unprecedented Membrane Morphology. Viruses. 7, 5686-5704 (2015).
- Paul-Gilloteaux, P., et al. eC-CLEM: flexible multidimensional registration software for correlative microscopies. Nature Methods. 14, 102-103 (2017).
- Polishchuk, R. S., Mironov, A. A. Correlative video light/electron microscopy. Current Protocols in Cell Biology Supplement. , 8 (2001).
- McDonald, K. L. A review of high-pressure freezing preparation techniques for correlative light and electron microscopy of the same cells and tissues. Journal of Microscopy. 235, 273-281 (2009).
- Newman, G. R., Jasani, B., Williams, E. D. A simple post-embedding system for the rapid demonstration of tissue antigens under the electron microscope. The Histochemical Journal. 15, 543-555 (1983).
- Schwarz, H., Humbel, B. M. Influence of fixatives and embedding media on immunolabelling of freeze-substituted cells. Scanning Microscopy. 3, 57-63 (1989).
- Luby-Phelps, K., Ning, G., Fogerty, J., Besharse, J. C. Visualization of identified GFP-expressing cells by light and electron microscopy. Journal of Histochemistry and Cytochemistry. 51, 271-274 (2003).
- Nixon, S. J., et al. A single method for cryofixation and correlative light, electron microscopy and tomography of zebrafish embryos. Traffic. 10, 131-136 (2009).
- McDonald, K. L. Rapid embedding methods into epoxy and LR White resins for morphological and immunological analysis of cryofixed biological specimens. Microscopy and Microanalysis. 20, 152-163 (2014).
- Webster, P., Schwarz, H., Griffiths, G. Preparation of cells and tissues for immuno EM. Methods in Cell Biology. 88, 45-58 (2008).
- Kukulski, W., et al. Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. Journal of Cell Biology. 192, 111-119 (2011).
- Peddie, C. J., et al. Correlative and integrated light and electron microscopy of in-resin GFP fluorescence, used to localise diacylglycerol in mammalian cells. Ultramicroscopy. 143, 3-14 (2014).
- Hampoelz, B., et al. Pre-assembled Nuclear Pores Insert into the Nuclear Envelope during Early Development. Cell. 166, 664-678 (2016).
- Lemercier, N., et al. Microtome-integrated microscope system for high sensitivity tracking of in-resin fluorescence in blocks and ultrathin sections for correlative microscopy. Scientific Reports. 7, 13583 (2017).
- Takizawa, T., Powell, R. D., Hainfeld, J. F., Robinson, J. M. FluoroNanogold: an important probe for correlative microscopy. Journal of Biological Chemistry. 8, 129-142 (2015).
- Romero-Brey, I., Yamauchi, Y. 3D electron microscopy (EM) and correlative light electron microscopy (CLEM) methods to study virus-host interactions. Methods in Molecular Biology: Influenza Virus Methods & Protocols. , (2018).
