STORM achieves super-resolution imaging by activating individual photoswitchable fluorophores stochastically. The location of every fluorophore is recorded and a super-resolution image is then constructed based on these locations4. Therefore, the precision of the fluorophore location is important for the super-resolution image reconstruction. The absorption spectra of Prochlorococcus peak at 447 nm and 680 nm,and Prochlorococcus has a minimum absorption at wavelengths above 700 nm16. However, Prochlorococcus MED4 cells still emitted high autofluorescence when exposed to an extremely high intensity of the 750-nm laser (Figure 3A), which is required for STORM imaging. Thus, the high autofluorescence background of Prochlorococcus cells heavily interferes with super-resolution imaging.
In order to utilize STORM to investigate protein organizations in Prochlorococcus, we developed a photobleaching method. After exposure to a white light of high intensity for 30 min, the autofluorescence of Prochlorococcus MED4 cells decreased (Figure 3B), although several cells with autofluorescence were still detected. We further elongated the photobleaching time to 60 min and found that the majority of the cells lost their autofluorescence (Figure 3C). These results indicate that the photobleaching method we developed here can greatly decrease the autofluorescence in photosynthetic organisms.
After photobleaching, we were able to use STORM to visualize the cell division protein FtsZ in Prochlorococcus MED4 cells. The cyanobacterium Prochlorococcus is the smallest and the most abundant photosynthetic organism on earth17. The diameter of a Prochlorococcus cell is only 500 – 700 nm18. With such a small cell size, it is impossible to visualize the FtsZ ring organization in Prochlorococcus cells using conventional wide-field fluorescence microscopy (Figure 2A). However, STORM has a spatial resolution of 9.6 nm in the xy-plane and 41.6 nm in the z-axis. Using STORM, we were able to reveal a detailed morphology of the FtsZ ring in Prochlorococcus (Figures 2B and 4). By rotating 3-D STORM images, we identified four different types of FtsZ ring morphologies: clusters (Figure 4A, Movie S1), an incomplete ring (Figure 4B, Movie S2), a complete ring (Figure 4C, Movie S3), and double rings (Figure 4D, Movie S4). Gaps were observed in the complete FtsZ rings of Prochlorococcus (Figure 4C, Movie S3), which is similar to that found in Caulobacter cresentus19. These four types of FtsZ ring morphologies showed the assembly process of the FtsZ ring during the cell cycle of Prochlorococcus, and this study helped us to understand the role of the FtsZ ring during the cell division of Prochlorococcus9.
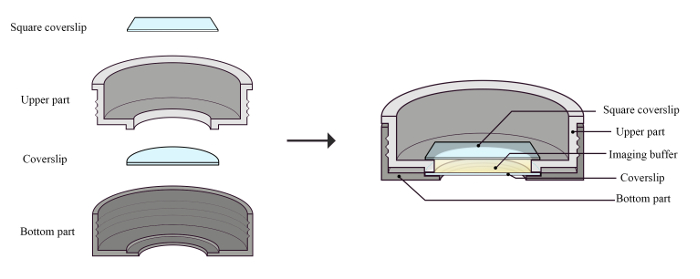
Figure 1: Components of the loading chamber and the assembled chamber with the coverslips and imaging buffer for STORM imaging. The coverslip with the sample was placed on the groove of the bottom part first. The upper part was then carefully screwed on to the bottom part. The imaging buffer was added in the loading chamber and then carefully covered with a square coverslip. Bubbles should be avoided to minimize any movement that may influence the accuracy of the measurement. Please click here to view a larger version of this figure.
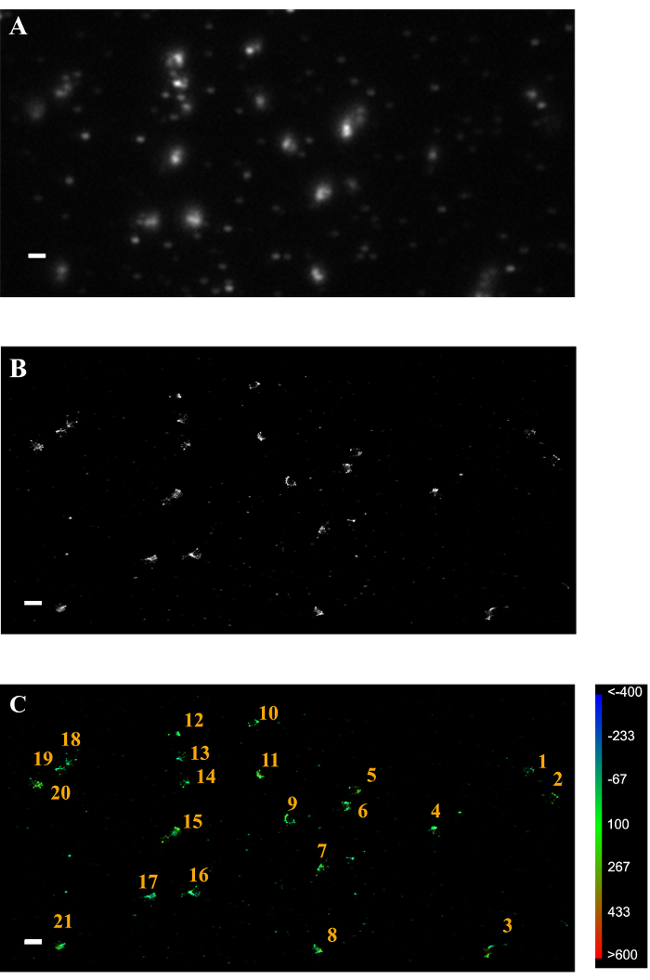
Figure 2: Representative wide-field, 2-D STORM, and 3-D color STORM images of FtsZ in Prochlorococcus MED4. The images were taken from the same field of view using regular (A) wide-field microscopy, (B) 2-D STORM, and (C) 3-D color STORM. The colors in panel C indicate the depth of the fluorescent signals on the z-axis. The numbers in panel C indicate the cells of interest. The scale bars = 1 µm. Please click here to view a larger version of this figure.
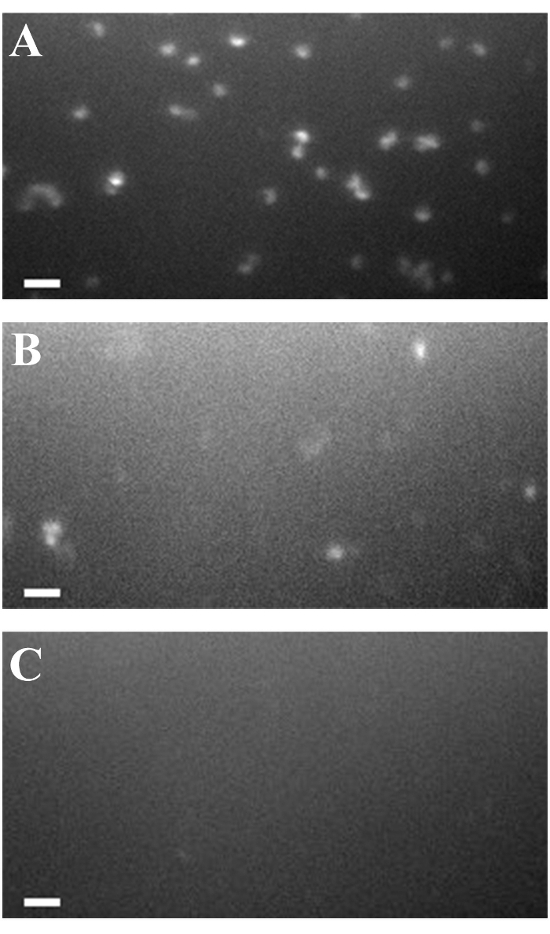
Figure 3: Photobleaching of Prochlorococcus MED4. Fixed Prochlorococcus MED4 cells were (A) not photobleached, or they were photobleached for (B) 30 min and (C) 60 min. The XD-300 xenon light was used at an intensity of 1,800 µmol/m2·s. Cells were excited by a 750-nm laser at the same intensity as was used for STORM imaging. The autofluorescences were imaged with the same filters as used in STORM to make sure the presence of minimal autofluorescence to affect STORM. The scale bars = 2 µm. Please click here to view a larger version of this figure.
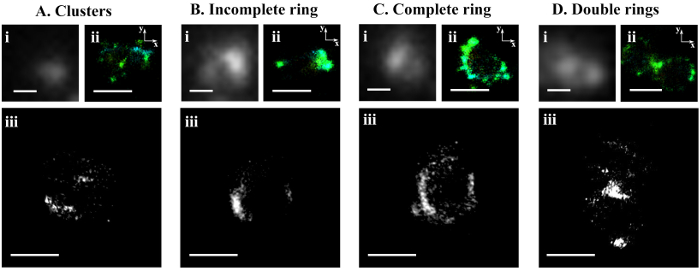
Figure 4: Representative STORM images of four FtsZ ring morphologies. Four different FtsZ ring morphologies were observed in Prochlorococcus: (A) clusters, (B) an incomplete ring, (C) a complete ring, and (D) double rings. For the same cell, images of (i) wide-field fluorescence microscopy, (ii) STORM on the xy-plane, and (iii) STORM after rotation were shown. The 3-D movies corresponding to the cells in panels A, B, C, and D are shown in Movies S1, S2, S3, and S4, respectively. The scale bars = 500 nm. Please click here to view a larger version of this figure.
| Chemicals | Stock solution | Final concentration |
| glucose | N/A | 10% (w/v) |
| Tris-Cl (pH 8.0) | 0.5 M | 50 mM |
| Ascorbic acid | 100 mM | 1 mM |
| Methyl viologen | 100 mM | 1 mM |
| Cyclooctatetraene | 200 mM | 2 mM |
| TCEP | 0.5 M | 25 mM |
| Glucose oxidase | 56 mg/ml | 0.56 mg/ml |
| Catalase | 4 mg/ml | 40 µg/ml |
Table 1: Recipe for the imaging buffer.
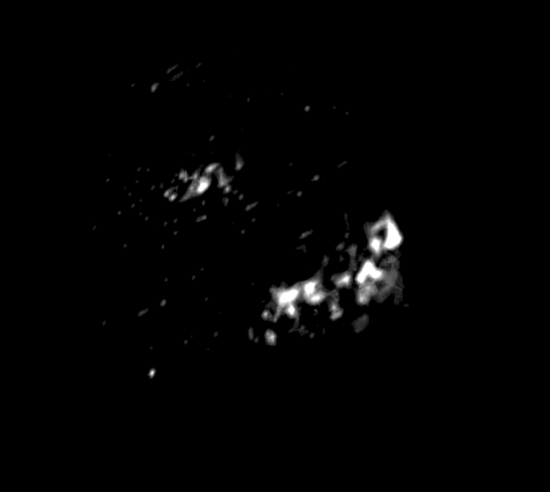
Movie S1: Clusters of FtsZ proteins observed in Prochlorococcus MED4 cells. Please click here to view this video. (Right-click to download.)
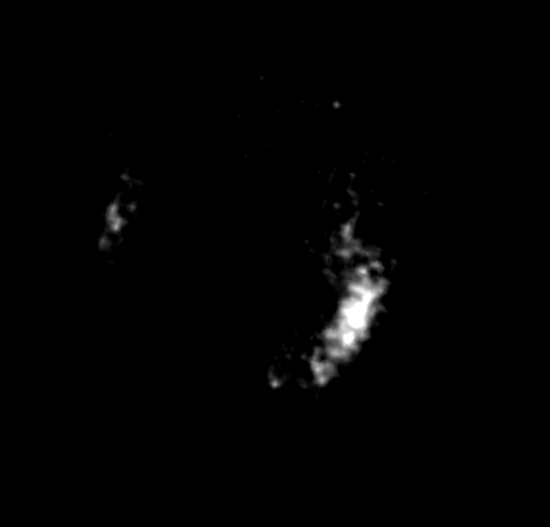
Movie S2: An incomplete ring of FtsZ proteins observed in Prochlorococcus MED4 cells. Please click here to view this video. (Right-click to download.)
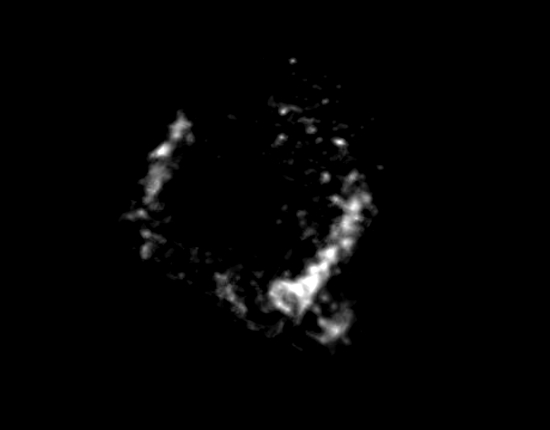
Movie S3: A complete ring of FtsZ proteins observed in Prochlorococcus MED4 cells. Please click here to view this video. (Right-click to download.)

Movie S4: A double-ring of FtsZ proteins observed in Prochlorococcus MED4 cells. Please click here to view this video. (Right-click to download.)
