In order to show the potential of SD-TIRF imaging, an assay was developed that should reveal the spatio-temporal organization of cell-matrix adhesion complexes and their interaction with the cytoskeleton during cellular adhesion. Therefore, adherent HeLa or, alternatively, NIH3T3 cells were transfected with YFP-Vinculin and RFP-Lifeact for 18-24 h, trypsinized and seeded onto fibronectin-coated glass bottom dishes. These cell lines were chosen for their pronounced cytoskeleton and higher robustness in live imaging experiments opposed to, for example, primary cells. Those might not withstand imaging in very sensitive condition as they are after trypsin treatment. At the microscope, YFP/RFP-expressing cells were selected and the adhesion process observed during a 60min time-lapse (Figure 2 and Movies 1 and 2). This specific assay has rarely and not clearly been described in the literature17,18. Moreover, adhesion formation has mostly been investigated in e.g. migrating cells19,20. Thus, we needed to adapt this methodology (cell line, coating, medium, composition) in order to carry out the experiments described in this paper.
As expected, cells were round-shaped at the beginning and only weakly adherent, whereas membrane protrusions were sensing the environment and making contact with the substrate. Cell-matrix contacts strengthened quickly upon formation of so-called nascent adhesions20,21 (Figure 2A, TIRF-488 channel, time points 0-4.5 min). The latter are spot-like, Vinculin-positive structures at the ventral side of the cell. The structures were clearly visible in the TIRF images. In the beginning of adhesion formation, actin was evenly distributed in the cell and did not localize to these early complexes (Figure 2A, SD-561 channel). Over the course of time, adhesion complexes enlarged and matured to focal adhesions (Figure 2C). These elongated structures were predominantly apparent at the periphery of the cell (Figures 2A+B) and resulted from forces that were exerted by acto-myosin fibers. These fibers started to connect to the adhesion complexes, thereby pulling them towards the cell center and inducing the strengthening of cell-matrix adhesions as well as the bundling of actin fibers21. Apparently, the cell also flattened as a result of actin network formation (Figure 2A, XZ view). SD-imaging proofed to be the method of choice here, as it allowed visualizing this process with high sensitivity, spatial resolution and from a complete perspective. In a previous report17, TIRF alone could only let speculate about the origin of peripheral adhesions, whereas SD-TIRF imaging clearly revealed its association with filopodia (Figure 2B, time point 17 min, white arrowhead). Indeed, actin fibers emerging centripetally from focal adhesions became visible after 27 min (Figure 2B, yellow arrowhead).
However, the acquisition settings of these experiments, i.e. acquisition speed, excitation intensity and detector gain, need to be carefully evaluated. The interval of 30 s/timepoint, enabling multi point acquisition, appears to be ideal, while the radiation intensity of the excitation laser (between 0.5-1 W/cm²) needs to be taken under critical consideration. Figure 2D displays a cell at a different position in the same experiment that failed to attach to the substrate. It might be possible that phototoxic effects affected the biology of the cell here, which finally resulted in membrane bubbling after circa 60 min, probably resembling apoptosis (Movie 2). This made again clear how sensitive cells can react to phototoxicity and that it is important to find a good balance between the amount of light put in and the information being taken out. Reducing the laser power, the number of images in a z-stack or increasing the gain might be the correct strategy for reducing phototoxicity. All these settings, however, should be adjusted at a level that still allows achieving enough resolution and signal to noise ratio, enabling to extract quantitative information from the recorded time lapse.
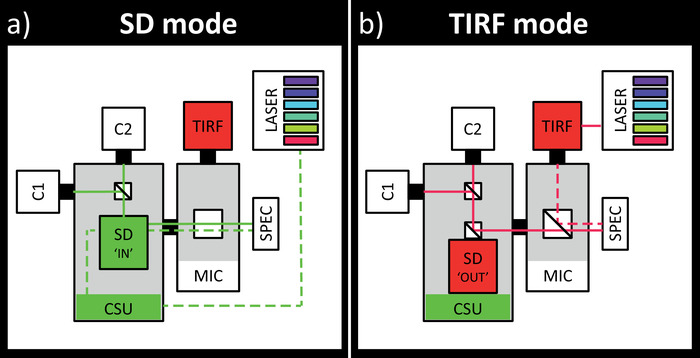
Figure 1: Schematic drawing of the SD-TIRF setup. A. SD imaging mode: the 6 different laser lines (405/445/488/515/561/640nm, green lines) are coupled into the confocal scanning unit (CSU), passing through the SD (position 'IN') and an empty filter cube in the microscope body (MIC). Fluorescence emission from the specimen (SPEC) is projected through the pinholes of the SD and split by different dichroic mirrors, e.g. for green/red or yellow/cyan simultaneous acquisition, onto two different EM-CCD cameras (C1 and C2). Fluorescence filters are placed in front of the cameras (not shown). B. TIRF imaging mode: the laser lines are coupled into the TIRF scanner (TIRF). A multi-line beam splitter in the MIC directs the beam to the specimen and the emission light bypasses the SD ('OUT') for maximized transmission. Fluorescence is detected by the same cameras and emission filters described in A. This figure has been modified from Zobiak et al.7 Please click here to view a larger version of this figure.
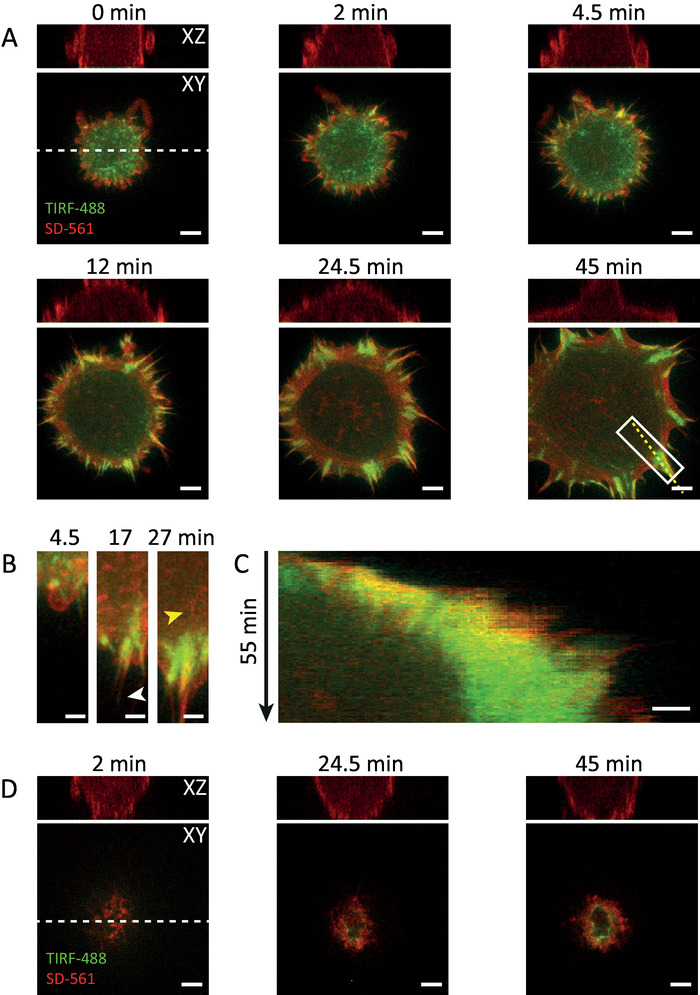
Figure 2: Representative results of cell spreading and adhesion formation using SD-TIRF microscopy. A. Double-transfected HeLa cell expressing RFP-Lifeact (red, SD-561 channel) and YFP-Vinculin (green, TIRF-488 channel) were trypsinized and re-seeded onto fibronectin-coated glass coverslips. Adhesion formation becomes readily visible, starting with small nascent adhesions (Vinculin-positive spots) at 0 minutes that develop into larger focal adhesions after circa 5 minutes. Cortical actin is apparent after circa 10 minutes and extends at the periphery of the cells (see frames at 12 and 24.5 minutes). B. The magnified view of the boxed region in A (frame at 45 minutes) depicts cell spreading and the transition from nascent to focal adhesions as well as filopodia-associated adhesions (white arrowhead) and stress fiber formation (see frame at 27 minutes, yellow arrowhead). C. Kymograph of the dashed yellow line drawn in A. D. (Photo-) Toxic effects on cells or otherwise unhealthy cells can let them fail to attach to the substrate. Imaging conditions have to be critically evaluated in order to exclude phototoxicity. Scale bar = 5 µm (in A and D) and 2 µm (in B and C). The XZ views in A and D are the orthogonal projections extracted from the dashed white lines drawn therein (bottom = substrate). Images were linearly contrast-enhanced and median-filtered with a 3×3 kernel. Please click here to view a larger version of this figure.
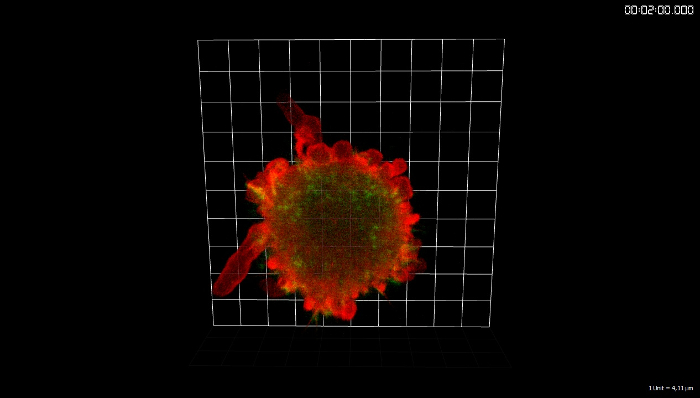
Movie 1: 3D-reconstruction of the timelapse sequence in Fig. 2A. The image sequence was rendered with 3D imaging software. Duration: 42.5 min. Acquisition rate: 2 dual-channel SD-TIRF stacks per minute (56 frames in total) were acquired. Please click here to view this video. (Right-click to download.)
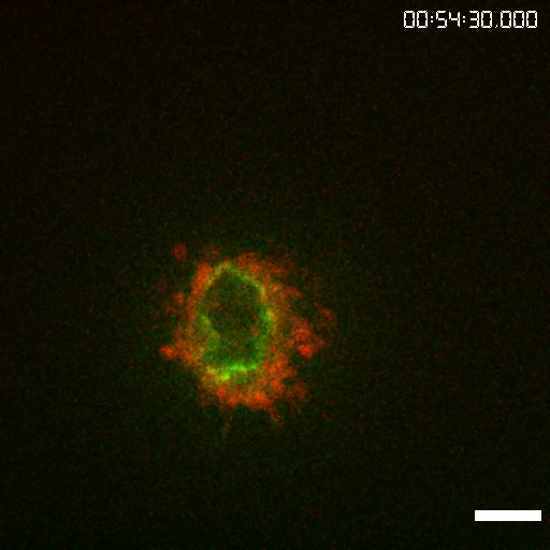
Movie 2: Movie of the timelapse sequence in Fig. 2C. Duration: 60 min. Acquisition rate: 2 dual-channel SD-TIRF stacks per minute (56 frames in total) were acquired. Please click here to view this video. (Right-click to download.)
