This method covers a multi-step process to isolate antigen-specific antibodies from human donors. In the representative data shown here, cells were incubated with a pool of fluorescently-labeled peptides representing several different domains of the tau protein, including phosphorylated peptides to mimic putative phosphorylation sites. These peptides were used as "bait" to identify cells that are reactive with tau epitopes(s) of interest. In preparation for sorting, a panel of fluorescently-labeled phenotypic markers were used to identify different cell populations within the enriched B cell population. A series of cytometry gates were devised to isolate the target memory B cells (Figure 1). Lymphocytes were isolated based on their cell size and granularity using forward scatter (FSC) and side scatter (SSC) plots in flow cytometry6,7. Following exclusion of multiple cells ("doublets") and dead cells, phenotypic markers allowed the segregation of IgG+ memory B cells via IgG, CD19 (B cell) and CD27 (memory). In this approach, the CD27 marker does not distribute into two discrete populations, so the top 45% of CD27+ expressing cells are included for the final gate. Finally, cells double-positive for both APC and PE fluorophores distribute into the upper right quadrant of the gating graph, indicating reactivity to both labeled versions of peptides. The cells that fell within the drawn gate were isolated and sorted into individual wells of a 96-well plate. The use of antigen with two different labels increases the signal-to-noise ratio and reduces the number of false positives in the subsequent molecular biology process. The sorted cells represented about 0.1% of the memory B cells in the final gate, and 0.001% of the starting cell sample.
The first readout of single cell cloning is confirmation of amplification of the respective heavy and light variable chains (Figure 2). Since paired recovery of both amplicons is desired, the PCR reactions are evaluated side-by-side on agarose gel, and matched pairs are excised from the gel and DNA fragments extracted. Typical efficiency of amplification is 30-50% heavy chain and 50-70% light (kappa) chain. Recovery of paired amplicons is usually between 25-40% efficiency. These efficiencies vary between donor pools, and the representative data (Figure 3) is an example of very efficient amplification from 24 single cells (42% paired recovery). Following IgG cloning, the IgG expression vector is transfected into human embryonic kidney (HEK-293) suspension cells, which are used to maximize expression. The use of serum-free medium reduces contaminating proteins from the recombinant antibody prep, and helps minimize noise in subsequent binding assays. The recovered antibodies are screened against the original panel of tau peptides and scored for reactivity by ELISA (Figure 4). An initial threshold of OD = 0.5 above background is used to indicate positive antigen reactivity, and β-actin protein is used as a control for non-specific binding. If the flow cytometry and sorting steps use a mixed pool of peptides, the screening ELISA is the first step of deconvoluting specific reactivities of the recovered IgGs. Three of the 10 IgGs assayed demonstrated reactivity against phosphorylated CBTAU22.1 peptide, and one was reactive to non-phosphorylated CBTAU27.1 (Figure 4A). Additional confirmation was completed using a concentration curve of the same recombinant antibody samples against the peptides identified in the initial screen, and an additional peptide as a negative control (Figure 4B). For each positive hit, an individual plasmid clone was isolated from the transformed pool and reconfirmed by the same ELISA method. Only Clone 34 is shown the data presented, and while reactivity to non-phosphorylated CBTAU27.1 was confirmed, lower affinity binding was also observed with the phosphorylated 27.1 peptide (Figure 4C).
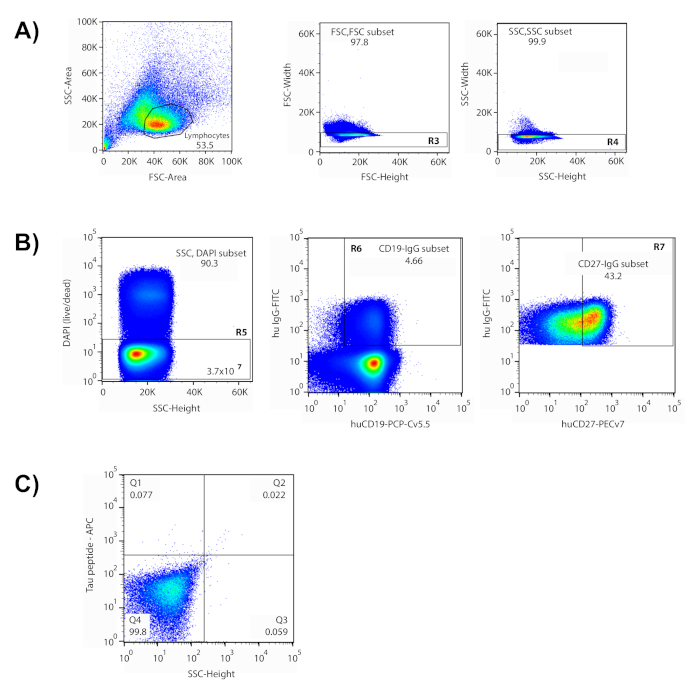
Figure 1: Isolation of antigen-reactive single cells via flow cytometry. (A) Shown is a representative plot where the lymphocyte population was contained within the drawn gate. (B) Gates based upon forward and side scatter (FSC-height v FSC-width (R3); SSC- height v SSC- width(R4)) were used to exclude doublets. Only cells within drawn gates were evaluated in subsequent plots. DAPI+ cells were considered dead and excluded (R5). In this experiment, 3.7 x 107 "live" cells were interrogated. The majority of live cells were B cells (CD19+), and IgG+ cells (4.66%) were isolated from this population (R6). The top 43.2% of CD27+-expressing memory cells were included in the selection gate (R7). (C) Quadrant 2 (Q2) contained cells reactive to both labeled antigens (peptide-APC v peptide-PE), and these cells were sorted and recovered individually into a 96-well plate. Please click here to view a larger version of this figure.
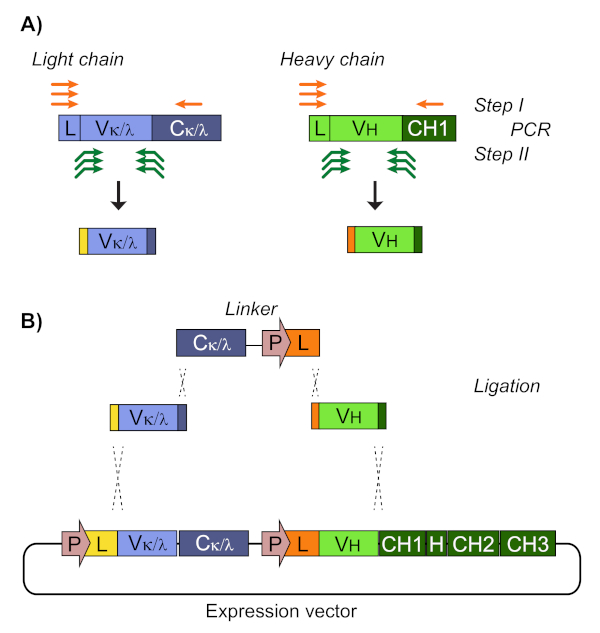
Figure 2: PCR amplification recovers IgG heavy and light variable sequence for cloning. (A) Both heavy (VH) and light (Vk or Vl) chain variable regions are recovered using nested PCR reactions. Step I primers amplify forward from the native leader sequence and reverse from within the constant region. Multiple arrows represent a pool of between 7-12 primers, which are used to ensure broad coverage of possible germlines. Step II primers are nested, specific to the extreme ends of the variable open reading frame, and add sequence to the ends of the amplicons that are homologous to adjacent sequence in the expression vector. (B) The linker consists of the constant light chain (Ck or Cl), followed by the heavy chain promoter and a non-native signal peptide sequence. The amplicons, linker, and plasmid backbone are simultaneously ligated via overlapping homologous sequence generated during the Step II PCR amplification, and isolated as an intact plasmid. The recombinant heavy and light chains in the final expression vector are driven by independent (and identical) CMV promoters, and are translated as separate proteins. Please click here to view a larger version of this figure.
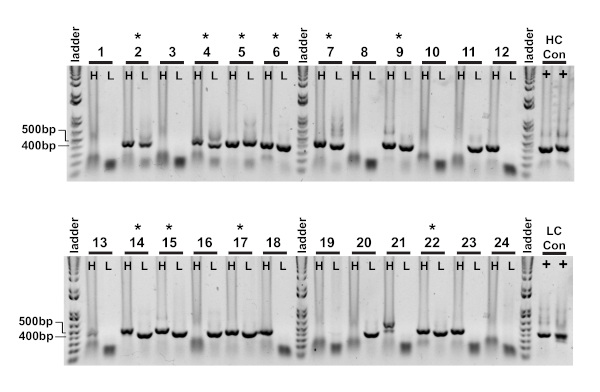
Figure 3: IgG heavy and light variable chain amplicons are recovered from single cells. The nested PCR reactions (Step II) were directly loaded onto 1% agarose gel and visualized. Heavy chain (H) and kappa light chain (L) PCR reactions from the same cell were loaded in adjacent wells so paired amplicons were easily observed. Successful heavy and light chain products are roughly 400 bp and 350 bp respectively. Successful recovery of paired amplicons are denoted by an (*). Please click here to view a larger version of this figure.
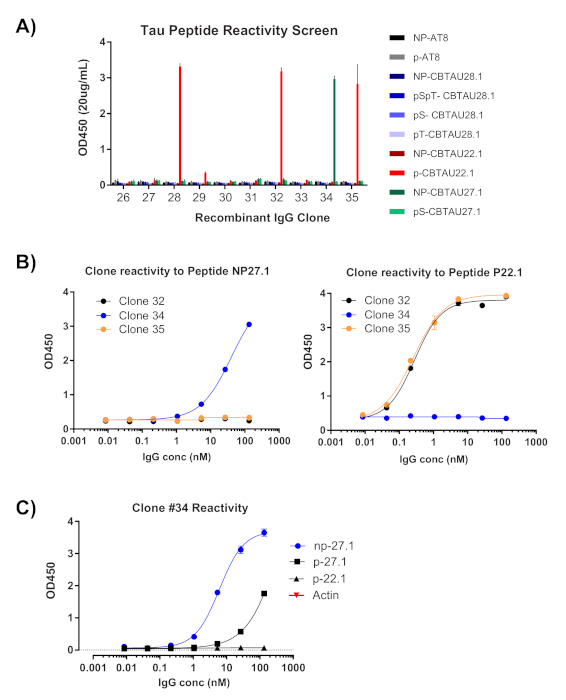
Figure 4: Antigen specificity is confirmed from recombinant IgGs. Plasmids containing the recovered heavy and light chain sequences are transfected into HEK cells for expression. After four days, recombinant antibodies are assessed for reactivity by ELISA. (A) IgG concentration in the clarified media was measured and diluted to 20 µg/mL. The pool of peptides used for sorting were individually assessed using 40 pmol per well in streptavidin plates. All IgGs were tested in duplicate wells. (B) Clones that displayed OD450 measurement >0.5 were re-assessed (Clones #32, #34, #35) using 1:5 dilution steps against the peptides identified in the screen. (C) Once confirmed as a hit, a single plasmid clone was isolated from the clone #34 transformed ligations, transfected, and reconfirmed by ELISA. The same was done for clones #32 and #35 (data not shown). Please click here to view a larger version of this figure.
