Wir beschreiben einen Bildansatz der totalen inneren Reflexionfluoreszenz-Anzahl und Helligkeit (TIRF-N&B) zur Bestimmung des durchschnittlichen oligomeren Zustands von Rezeptormolekülen an der Plasmamembran lebender Zellen mit dem Ziel, die Rezeptor-Baugruppe zu verknüpfen. Dynamik zur biologischen Funktion der Proteine (Abbildung 1).
Bei extrazellulärer Ligandenbindung initiieren Rezeptoren die intrazelluläre Signaltransduktion in Abhängigkeit von ihrer Konformation, Oligomerisierung, potenziellen Co-Rezeptoren und Membranzusammensetzung. Trotz der Bedeutung und Allgegenwart der Rezeptor-Oligomerisierung, anerkannt als schlüsseleines Ereignis in der zellulären Signalisierung1,2,3,4,5,6, 7können nur wenige Methoden Clustering-Ereignisse erkennen und den Grad des Clusterings experimentell messen8,9. Das konfokale Volumen (x,y bei 300 nm, z x 900 nm) ist für den Nachweis der molekularen Interaktion und Stoichiometrie nicht ausreichend aufgelöst, auch nach der Optimierung durch Bildwiederherstellungsalgorithmen10. Die Untereinheitszusammensetzung von Proteinoligomeren kann nicht auch nur durch Superauflösungsmethoden bei einer Auflösung von 20-70 nm wie PALM11, STORM12und STED13rein räumlich gelöst werden. Darüber hinaus kann ihre zeitliche Auflösung (in der Reihenfolge von Minuten pro Bild) der Kinetik im Sekundenbereich nicht folgen. Einzelne Molekül-Stepbleichung löst die Stoichiometrie von Protein-Oligomeren nur, wenn sie unbeweglich sind14.
Eine der vielseitigsten Methoden zur Messung der Dichte und Oligomerisierung fluoreszierender Proteine in Einzelbildern ist die räumliche Intensitätsverteilungsanalyse (SpIDA), die auf räumlicher Probenahme beruht. Es ist sowohl auf chemisch fixierte als auch auf lebende Zellen anwendbar und ermöglicht die gleichzeitige Analyse mehrerer Interessenbereiche der Zelle mit Standardfluoreszenzmikroskopie15. Alternativ eignen sich Momentmethoden wie Fluoreszenz-Korrelationsspektroskopie (FCS)16, Photonenzählhistogramm (PCH)17und Zahl und Helligkeit (N&B)18,19, für quantitative oligomere Messungen. Diese Methoden analysieren die Fluoreszenzintensitätsschwankungen, die in der Zeit beobachtet werden können, wenn die Fluorophore in und aus einem Beleuchtungsvolumen diffundieren. Die Amplituden der Intensitätsschwankungen können eindeutig durch die molekulare Helligkeit des Fluorophors () und die durchschnittliche Anzahl der Fluorophore (n) innerhalb des Beleuchtungsvolumens17 (Abbildung 2) beschrieben werden. Typischerweise können der Diffusionskoeffizient der Fluorophore und die durchschnittliche Anzahl der Moleküle (umgekehrt bezogen auf den G(0)-Wert) innerhalb des Beleuchtungsvolumens durch FCS20ermittelt werden. Da die Diffusionszeit jedoch nur mit der kubischen Wurzel der Masse skaliert, ist FCS nicht empfindlich genug, um Veränderungen in der Molekularmasse21zu erkennen. In der Praxis kann einfarbiges FCS die Dimerisierung von Membranrezeptoren nicht erkennen. PCH löst Mischungen verschiedener Oligomere genau auf. Mit mehr als zwei Momenten der Amplitudenverteilung erkennt es Moleküle unterschiedlicher Helligkeit, die das gleiche Beleuchtungsvolumen belegen. Scannen von FCS22 und Entwicklungen, wie z. B. der interessante Paarkorrelationsansatz der molekularen Helligkeit (pCOMB)23, eingeführt, um den Alikatbereich der Anwendbarkeit von Fluoreszenzkorrelationsmethoden in biologischen Systemen zu erweitern24 bleiben Einzelpunktmethoden, denen die Fähigkeit zu schnellen Messungen in einem großen Zellbereich fehlt, was viele aufeinander folgende Beobachtungen an jedem Pixel und die Datenerfassung in der Reihenfolge von Sekunden erfordert.
N&B ist eine vereinfachte Version von PCH, die nur den ersten und zweiten Moment der Amplitude der Fluoreszenzverteilung berücksichtigt, nämlich die mittlere Intensität, , und die Varianz,2 (Abbildung 2)18,19 und aus diesem Grund kann sie den mollaren Anteil unbekannter Oligomere in einer Mischung nicht bestimmen, sondern schätzt nur den durchschnittlichen Oligomerisierungszustand der Mischung. Dennoch hat N&B den Vorteil, mit relativ kleineren Zeitreihen von Bildern von Lebendenzellen als PCH pixelweise zu arbeiten, indem einfach die Zeitlichen schwankungen der Fluoreszenzintensität überwacht werden. Da N&B die Zeit pro Pixel auf wenige Mikrosekunden reduziert, kann es einer schnellen Oligomerisierungskinetik über große Zellbereiche folgen und eine Bildaufnahme auf einer Zeitskala von Sekunden in der Raster-Scanmikroskopie (z. B. konfokale, 2-photon) und Millisekunden ermöglichen. in der kamerabasierten Mikroskopie (z.B. TIRFM).
Mehrere Berichte haben die Fähigkeit von N&B gezeigt, die Anzahl der Untereinheiten in Proteinclustern zu quantifizieren, indem erweiterte Zellregionen geabbildungen. Paxillin-EGFP-Cluster wurden an den Haftstellen in den CHO-K1-Zellen25nachgewiesen und die intrazelluläre Aggregation des pathogenen Httex1p-Peptids in COS-7-Zellen26beschrieben. N&B wurde nach der Liganden-gesteuerten Oligomerisierung des ErbB-Rezeptors27und der Wirkung des Liganden FGF21 auf Klothob (KLB) und FGFR1c in den HeLa-Zellen28angewendet. Die Kombination von TIRF-Bildgebung und N&B-Analyse wurde verwendet, um zu zeigen, dass Dynamin-2 in erster Linie tetramerisch in der gesamten Zellmembran29ist. Wir haben N&B sowohl auf Raster-Scanning als auch auf TIRF-Bilder angewendet, um die Liganden-gesteuerte Dimerisierung von uPAR- und FGFR1-Zellmembranrezeptoren30,31zu beweisen.
Fluoreszenzkorrelationsmethoden wie N&B, FCS und PCH basieren auf der Vorstellung, dass in einem offenen Volumen die Besatzungszahl der Teilchen einer Poisson-Verteilung folgt. Da nur die Photonen, die die Fluorophore emittieren, erkannt werden können, ist der Mittelwert  für eine gemessene Fluoreszenzintensität im Vergleich zur Zeit in einem Pixel des Bildes, das Produkt der durchschnittlichen Anzahl von Fluorophoren im Beleuchtungsvolumen, n, und molekulare Helligkeit, Nr.17:
für eine gemessene Fluoreszenzintensität im Vergleich zur Zeit in einem Pixel des Bildes, das Produkt der durchschnittlichen Anzahl von Fluorophoren im Beleuchtungsvolumen, n, und molekulare Helligkeit, Nr.17:

wobei die Anzahl der Photonen, die pro Zeiteinheit (konventionell pro Sekunde) pro Molekül emittiert werden, wenn sich das Molekül im Zentrum des Beleuchtungsvolumens befindet.
Helligkeit ist eine Eigenschaft jedes Fluorophors in einer bestimmten Anschaffung eingerichtet, während Intensität ist die Summe aller Beiträge aus allen Fluorophoren. In biologischen Wettbewerben wird die Helligkeit mit der Zunahme der Anzahl der Fluorophore, die zusammen schwanken, zunehmen, was Informationen über den Oligomerisierungszustand des fluoreszierend getaggten Proteins gibt. Die Fluktuationsamplituden an einem bestimmten Pixel werden anhand der Varianz des Fluoreszenzsignals gemessen,2:

Wobei der Mittelwert des Quadrats der Intensität  , und das
, und das  Quadrat des Mittelwerts der Intensität , aus den individuellen Intensitätswerten in jedem Pixel jedes Frames berechnet werden:
Quadrat des Mittelwerts der Intensität , aus den individuellen Intensitätswerten in jedem Pixel jedes Frames berechnet werden:
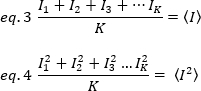
wobei K die Anzahl der Gesamtframes in der Zeitreihe ist. Experimentell ist es notwendig, für die gesamte Bildreihe die Varianz zu berechnen, die die Streuung der einzelnen Intensitätswerte an jedem Pixel eines einzelnen Bildes um den mittleren Intensitätswert beschreibt. Die Varianz umfasst alle Schwankungen unterschiedlicher Herkunft. In einer ersten Annäherung kann die Varianz durch die diffundierenden Partikel im Beleuchtungsvolumen,20, durch das Detektor-Schussrauschen von der Varianz getrennt werden,2d. Die beiden Varianzen sind unabhängig; somit wird die Gesamtabweichung durch ihre Summe angegeben:

Die Varianz aufgrund molekularer Schwankungen im und aus dem Nachweisvolumen ist linear abhängig von der molekularen Helligkeit und Intensität:

Neuanordnung eq. 6 nach 1q. 1:

Nach dem typischen Konzept der Fluoreszenzkorrelationsspektroskopie besagt Gleichung 7, dass die Varianz aufgrund der Anzahl der Schwankungen vom Quadrat der Partikelhelligkeit abhängt.
Dann ist die Varianz aufgrund von Detektorschwankungen eine lineare Funktion der erfassten Intensität, unter der Annahme, dass der Detektor unterhalb seiner Sättigungsgrenze19betrieben wird:

Bei Photonenzähldetektoren a=1 und c=0 entspricht die Detektorvarianz also der durchschnittlichen Intensität:

Um diese Konzepte auf reale Messungen in lebenden Zellen anzuwenden, definieren Gratton und Kollegen18 die scheinbare Helligkeit B für jedes Pixel als Verhältnis der Varianz zur durchschnittlichen Intensität:

B ist der Parameter, der experimentell gemessen wird. In dieser Arbeit werden Zeitreihenbilder von FGFR1-Rezeptoren an der Plasmamembran von HeLa-Zellen durch die TIRF-Mikroskopie erfasst und die durchschnittliche scheinbare Helligkeit B wird durch die N&B-Analyse bestimmt. Dann, nach Zugabe von FGF2, werden aufeinander folgende Zeitreihen erfasst, um die Veränderungen in der Selbstmontage der Rezeptormoleküle in der Membranoberfläche nach der Stimulation des Rezeptors mit dem kanonischen Liganden zu verfolgen.
Da es sich bei dem Detektor des TIRF-Mikroskops jedoch um eine EMVD-Kamera handelt, muss der Ausdruck für die scheinbare Helligkeit als19geändert werden:

wobei der Offset der Intensitätsausgleich der Erkennungselektronik ist, der ein Merkmal der Detektoreinstellungen ist. Die Varianz und die durchschnittliche Intensität eines analogen Detektors werden jeweils angegeben durch:
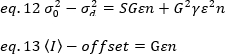
wobei G die analoge Verstärkung in digitalen Pegeln (DL/Photonen), S, die digitalen Pegel pro Photon19, durch die Steigung eines Intensitäts-Gegen-Varianzdiagramms für eine Lichtquelle mit konstanter Intensität (keine zeitlichen Schwankungen) angegeben wird. Der Faktor ist mit der Form des Pixelerkennungsvolumens verknüpft. Nach Hassler et al.32beträgt der Faktor 0,3 für die TIRF-Bildgebung, die mit der maximalen Verstärkung der Detektionskamera19arbeitet. Die Offset-, S- und G-Parameter sind Merkmale der Kamera und des Mikroskops. Die scheinbare Helligkeit B wird durch Neuanordnung von 11 nach 12 und 13 erreicht:

Experimentell ist die s eine komplexe Funktion der Laserintensität und der Detektionseffizienz des Systems. Da B/S jedoch linear von der Abhängigkeit von – abhängig ist, ist es nur wichtig, den relativen Wert von . für einen bestimmten Erkennungsmodus zu bestimmen:

wobei das S’ proportional zu . Dennoch wird eine Kalibrierung mit einem internen Verweis durchgeführt.
