Ventral fin wounding is followed by rapid neutrophil mobilization from the CHT into the ventral fin and clustering at the wound margin, within 30-60 min (Figure 1). We visualized the distribution of two chemokine receptors, Cxcr1 and Cxcr2, which are expressed by zebrafish neutrophils24 and recognize Cxcl8a and Cxcl8b14, using spinning-disk confocal microscopy. We generated two corresponding transgenic lines, Tg(lyz:Cxcr1-FT) and Tg(lyz:Cxcr2-FT), in which neutrophils express a fluorescent timer (FT) construct of the receptor, i.e. a fusion with a tandem of sfGFP and tagRFP (Figure 2 and reference14). The use of the two fluorophores was intended to allow monitoring of a broad range of receptor fates and provide estimates of protein turnover time at the plasma membrane, as newly synthesized receptors would fluoresce in green and progressively become red as they age8,14. However, these receptors were found to have fast constitutive turnover at the neutrophil plasma membrane and that the residence time was shorter than the maturation time of tagRFP, with sfGFP showing membrane localisation and tagRFP showing vesicular localization at steady state (Supplementary Video 1 and ref14). Therefore, we focused on the distribution of sfGFP to monitor ligand-induced internalization at sites of tissue damage. The pattern of receptor distribution was quantified using the contrast metric, which reports differences in intensity between neighboring pixels. The rationale is that when the receptor distribution is membranous and smooth, the contrast value is low. When the receptor distribution is vesicular and more punctate, then the contrast value is high (Figure 3).
An alternative method is to quantify the ratio of receptor levels (sfGFP intensity) over the levels of a control membrane marker e.g. membrane CFP (mCFP) (Figure 3). Both methods could detect receptor internalization, as indicated by more vesicular receptor distribution pattern globally in the cell (higher contrast value) or lower receptor levels at the membrane (lower sfGFP/mCFP ratio). However, the contrast metric could also detect receptor internalization in neutrophil clusters at the wound, in which membrane segmentation was less accurate and not applicable (Figure 3). Using this metric, we were able to quantify visible differences between Cxcr1 and Cxcr2 trafficking in neutrophils at wounds (Figure 4 and Supplementary Video 2). Cxcr1-FT internalized in cells located at the wound whereas Cxcr2-FT remained membranous in neutrophils at the wound (Figure 4A-C, Supplementary Video 2 and Supplementary Video 3). Suppression of Cxcl8a and Cxcl8b, through morpholino treatment, differentially affected Cxcr1-FT internalization at wounds (Figure 4C,D). To further validate that Cxcr1-FT responds to Cxcl8a, we performed chemokine response assays in early embryos. We found that Cxcr1-FT markedly internalized in embryos in which Cxcl8a was co-expressed (Figure 5). Altogether these results indicate that the described methods can be deployed to measure chemokine-induced receptor internalization in neutrophils and establish the identity of the ligand mediating these effects.
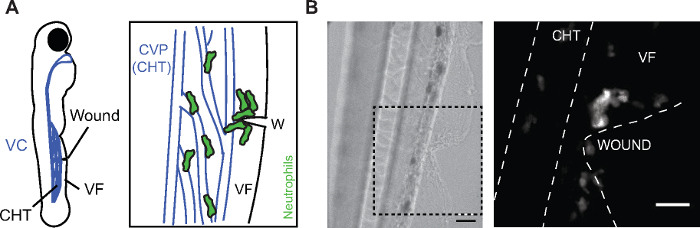
Figure 1: Neutrophil migration to ventral fin wounds. (A) (Left) Cartoon of 3 dpf larva showing the location of the caudal hematopoietic tissue (CHT), the venus circulation (VC, blue), the ventral fin (VF) and the wound site. (Right) Cartoon depicting the area of the wound (W) with neutrophils getting mobilized from the CHT and clustering at the wound. The caudal vein plexus (CVP) of the CHT tissue is drawn in blue. (B) Bright field image (left) and confocal projection (right) showing the ventral fin wound and the distribution of neutrophils in Tg(mpx:GFP) larvae at 2 h post-wounding. Dashed lines show VF and CHT outlines. Scale bar = 25 µm. Cartoon and fluorescent image modified from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
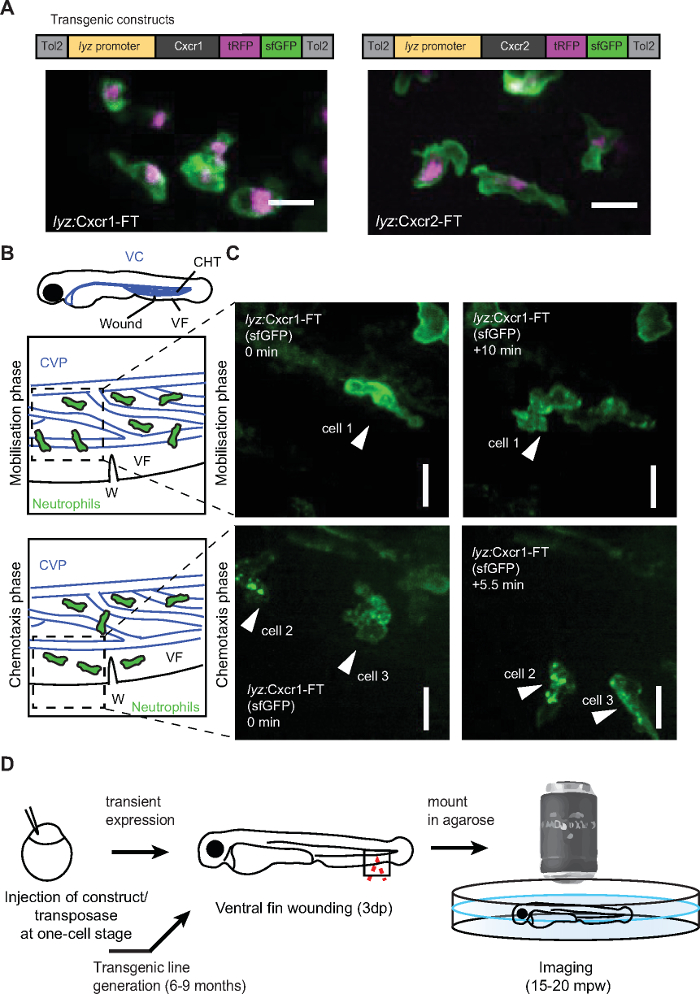
Figure 2: Live imaging of chemokine receptor trafficking in neutrophils. (A) Constructs used for neutrophil-specific transgenic expression of Cxcr1-FT (Fluorescent Timer) and Cxcr2-FT. Confocal projections of neutrophils in the head of a 3 dpf transgenic larva (Tg(lyz:Cxcr1-FT), top; Tg(lyz:Cxcr2-FT), bottom) showing tRFP (magenta) and sfGFP (green) channels. Scale bar = 20 µm. (B) Anatomical scheme of 3 dpf larva as in Figure 1A. Below the larva are schemes depicting the area of the wound (W) with neutrophils getting mobilized from the CHT (top) or performing chemotaxis upon entering the ventral fin (bottom). Dashed square indicates area imaged in snapshots on the right. (C) Neutrophils in Tg(lyz:Cxcr1-FT) larvae (sfGFP is shown) upon mobilization from the CHT (top panels) or chemotaxis towards the wound (bottom panels). Arrows show the same cells over time. Time points on the right image are minutes elapsed after image on the left. Scale bar = 10 µm. (D) Schematic representation of experimental approach for live imaging of chemokine receptor trafficking. Panels A-C modified from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
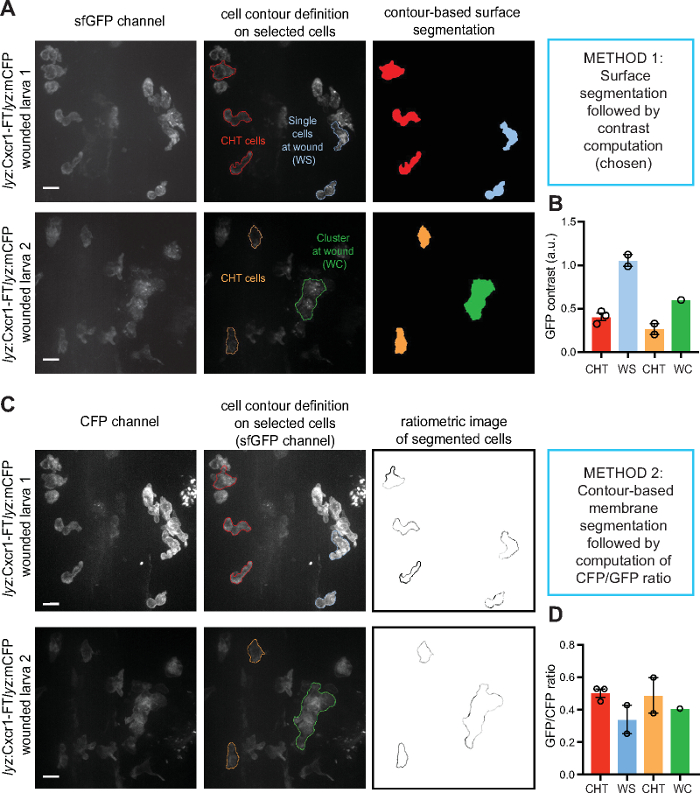
Figure 3: Quantification examples of receptor dynamics. Single (blue) or clustered neutrophils (green) at wounds or non-mobilized neutrophils in the CHT (red, orange) were segmented and analyzed by different methods to compare results. The same example cells shown were analyzed with two methods to relate what is seen in the image with the range of values extracted. (A) The surface of the selected, example cells were segmented based on contour definition in the sfGFP channel. (B) Contrast was computed from the example cells shown in A. (C) The membrane of the selected, example cells were segmented based on contour definition in the CFP channel. Ratiometric analysis of sfGFP/CFP followed. (D) The ratio of sfGFP/CFP was computed on the example cells shown in C. Error bars represent S.E.M. from individual cells, in cases of n>1, values here were not used for statistical analysis but merely to exemplify measurements obtained with the different quantification methods. Scale bar = 10 µm. Figure modified from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
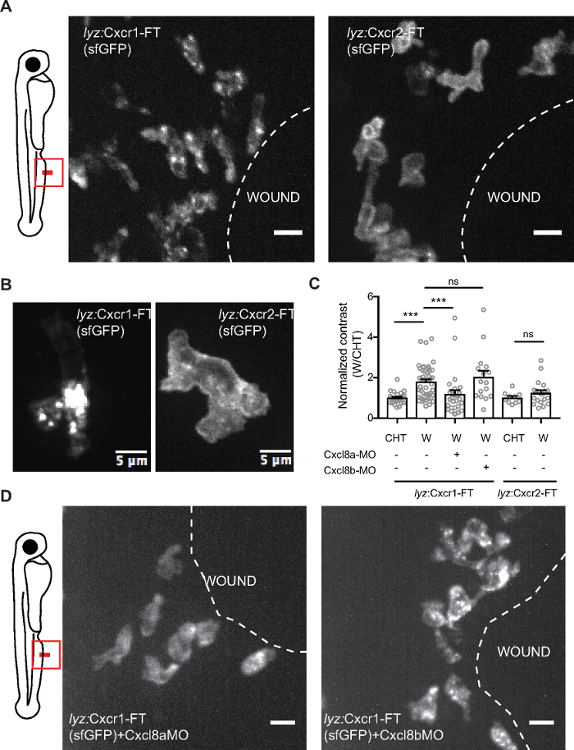
Figure 4: Differential dynamics of Cxcr1 and Cxcr2 in response to wounding. (A) Confocal projection of neutrophils in Tg(lyz:Cxcr1-FT) or Tg(lyz:Cxcr2-FT) larvae at the wound at 80 min post-wounding (sfGFP channel shown). Scale bar = 10 µm. (B) Magnified Cxcr1-FT neutrophil (left) and Cxcr2-FT (right) at the wound. Green receptor is shown in gray. Scale bar = 5 µm. (C) Normalized contrast (contrast per individual neutrophil normalized to the mean contrast of non-mobilized cells in the CHT). cxcl8a refers to injection of a splice-blocking together with a translation-blocking morpholino for cxcl8a. cxcl8b refers to injection with a splice-blocking morpholino for cxcl8b. For Tg(lyz:Cxcr1-FT): n=24 cells (CHT), n=47 cells (wound) from 8 larvae. For Tg(lyz:Cxcr1-FT) with morpholinos: n=28 cells (Cxcl8a-MO) from 5 larvae, n=16 cells (Cxcl8b-MO) from 5 larvae. For Tg(lyz:Cxcr2-FT): n=10 cells (CHT) and n=20 cells (wound) from 3 larvae. Data were pooled from independent larvae acquired in 1-5 imaging sessions. Kruskal-Wallis test with Dunn’s multiple comparisons test for Tg(lyz:Cxcr1-FT), two-tailed unpaired Mann-Whitney test for Tg(lyz:Cxcr2-FT). (D) Confocal projection of neutrophils in Tg(lyz:Cxcr1-FT) transgenic larvae treated with cxcl8a morpholino (MO) (left) and cxcl8b MO (right) responding to fin wounds (sfGFP channel shown in green). Snapshot taken at timepoints of equivalent neutrophil accumulation (85 min post-wounding in left image and 45 min post-wounding in right image). Scale bar = 10 µm. Figure modified from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
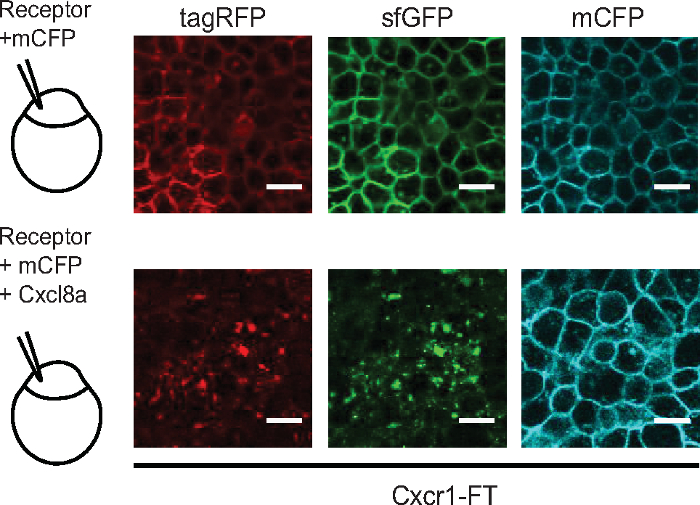
Figure 5: Chemokine response assay in early embryos. Laser-scanning confocal slices of gastrulating embryos showing expression and distribution of Cxcr1-FT. 100 pg of Cxcr1-FT mRNA was injected into one cell-stage eggs with or without 150 pg Cxcl8a mRNA. Green and red receptors are shown in separate channels. Control membrane CFP marker (mCFP) is shown in the cyan channel. Scale bar = 20 μm. Figure modified from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
Supplementary Movie 1: Transgenic neutrophils in the head of a Tg(lyz:Cxcr1-FT) (left) and Tg(lyz:Cxcr2-FT) (right) larva at 3 dpf. sfGFP(green), tagRFP (magenta). Frame interval is 30 sec and frame rate is 5 fps. Scale bar = 20 μm. Video originates from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to download this video.
Supplementary Movie 2: Neutrophils in Tg(lyz:Cxcr1-FT) (left) and Tg(lyz:Cxcr2-FT) (right) transgenic larvae responding to fin wounds. Movie starts within 10 min post-wounding and lasts 60 min. sfGFP (green), tagRFP (magenta). Frame interval is 30 sec and frame rate is 10 fps. CHT = caudal hematopoietic tissue. VF = ventral fin. Scale bar = 25 μm. Video originates from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to download this video.
Supplementary Movie 3. Additional examples of neutrophils from a wounded Tg(lyz:Cxcr1- FT) transgenic larva (different larva to that shown in Video 2), acquired at higher resolution, showing receptor internalization (sfGFP channel shown in green) upon mobilization in the CHT or upon entry and chemotaxis in the ventral fin. Frame interval is 30 sec and frame rate is 2 fps. Scale bar = 10 μm. Video originates from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to download this video.
Supplementary File 1: Please click here to download this file.
Supplementary File 2: Please click here to download this file.
Supplementary File 3: Please click here to download this file.
Supplementary File 4: Please click here to download this file.
