Our protocol describes an experimental framework for comparative fluorescence live imaging in sample chamber based LSFMs. For instance, the framework can be used to juxtapose (i) embryos of two or more species, (ii) embryos of lines in which one or more genes are knocked out plus wild-type controls, (iii) multiple embryos of the same transgenic line, (iv) embryos from different transgenic lines, or (v) embryos from sublines that carry the same transgene, but at different genomic locations. In this section, we provide examples for the last two scenarios.
In our first exemplary application, we show the fluorescence signal dynamics of three embryos that derive from different transgenic Drosophila lines (Table 2) over a period of about 1 day (Figure 4, Supplementary Movie 1). For these lines, we expected rather similar fluorescence patterns since all of them express nuclear-localized EGFP/GFP under control of different presumably ubiquitous and constitutively active promoters. However, our comparative live imaging results show that there are strong spatiotemporal differences in the expression patterns that are certainly not secondary effects due to ambient variance.
In our second application example, we compare the performance of three embryos that derive from the AGOC{Zen1’#O(LA)-mEmerald} #1, #2 and #3 (Zen1’LA-mE #1, #2 and #3, respectively) transgenic Tribolium sublines36. All of these carry the same piggyBac-based transgene that leads to expression of mEmerald-linked37 Lifeact38 under control of the zerknüllt 139 promoter, a transcription factor involved in serosa specification. Our comparative live imaging results, which illustrate the serosa window closure process during gastrulation, suggest that subline #2 provides a remarkably stronger overall signal than the other two sublines (Figure 5, Supplementary Movie 2). This indicates that also in Tribolium, the genomic context may have a strong influence on the expression level of transgenes that carry an expression cassette.
Successful retrieval of all six embryos that are shown in this section was possible as described in Step 8 of our protocol. All of them developed into fully functional and fertile adults (Supplementary Table 1, “Retrieval” row), indicating that the overall procedure, from dechorionation over mounting to recording, was non-invasive. This level of quality control is essential whenever wild-type development is expected, e.g., regarding control embryos that are imaged simultaneously with embryos in which one or more genes are knocked down or knocked out.
All datasets that were used to create the representative results are provided as resources that will help especially LSFM novices to evaluate the quality of their own work. The digital object identifier-based download links can be found in the metadata table (Supplementary Table 1, “Data Access” row).
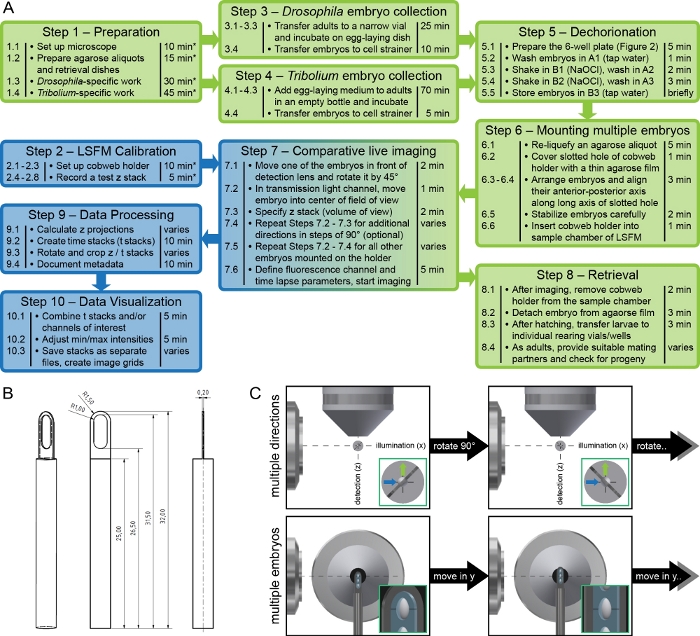
Figure 1: Experimental framework and mounting method overview. (A) Flowchart of the ten protocol steps with short reminders and an estimation of the required time. Tasks marked with asterisks do not have to be performed during every assay but depending on the circumstances. Green boxes indicate steps associated with Drosophila and/or Tribolium, blue boxes indicate steps associated with microscopy. (B) Detail drawing of the cobweb holder. The presented design is suitable for cylinder-based clamp mechanisms, which are commonly used in sample chamber-based LSFMs. Specifications are in millimeters. The holder can be scaled as long as the working distance of the detection lens is respected. (C) Recording sequence for live imaging of multiple embryos along multiple directions. At first, z stacks of the uppermost embryo are recorded along up to four orientations, i.e. 0°, 90°, 180° and 270°. Subsequently, the embryo below is moved in front of the detection lens and the recording/rotation sequence begins anew. Please click here to view a larger version of this figure.
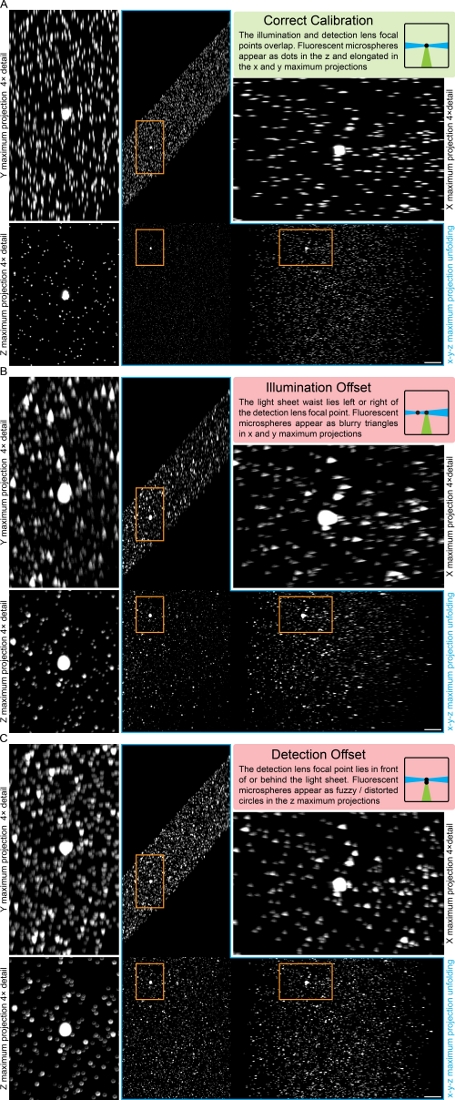
Figure 2: Fluorescent microsphere-based calibration chart for LSFMs. The chart illustrates how fluorescent microspheres appear in the x, y and z maximum projections if the LSFM is correctly calibrated (A), or if there is an illumination (B) or detection offset (C). Please click here to view a larger version of this figure.
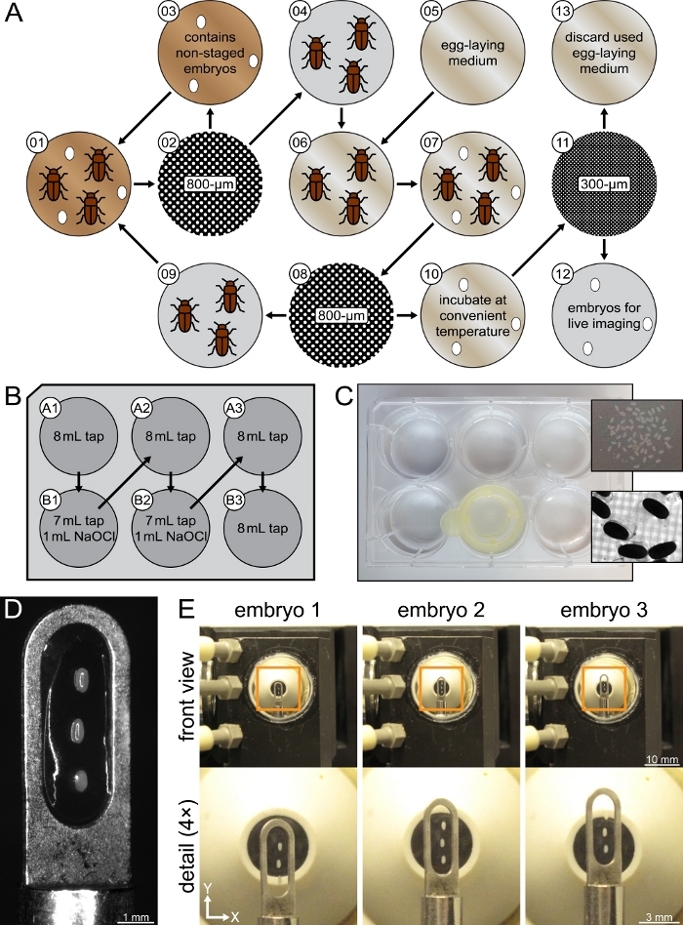
Figure 3: Collection, dechorionation, mounting and imaging of multiple embryos. (A) Tribolium embryo collection overview. The illustrations are cited throughout Step 4 according to the numbers in the upper left. (B) Preparation and transfer scheme for the 6-well plate used during Step 5. The B1 and B2 wells contained diluted sodium hypochlorite (NaOCl), which induces dechorionation. (C) The 100 µm cell strainer, which was used to transfer the embryos from one well to another, within the B2 well. The upper detail image shows a close-up of several Tribolium embryos on top of the cell strainer mesh, the lower detail image shows a transmission light stereo microscope image of several Tribolium embryos with partially detached chorion. (D) Cobweb holder with three mounted Tribolium embryos. The embryos, as well as their anterior-posterior axes, were aligned along the long axis of the slotted hole. (E) Movement of the cobweb holder within the sample chamber of the LSFM during the recording of three embryos. Please click here to view a larger version of this figure.

Figure 4: Comparative fluorescence live imaging of one embryo each from the #01691, #29724 and #24163 transgenic Drosophila lines. Embryos are shown along two (of four recorded) directions over a period of 20:00 h (from a total imaging period of 23:40 h). No dynamic intensity correction over time was performed. Metadata for the Drosophila datasets can be found in Supplementary Table 1. ZA, Z maximum projection with image adjustment. Scale bar, 100 µm. Please click here to view a larger version of this figure.
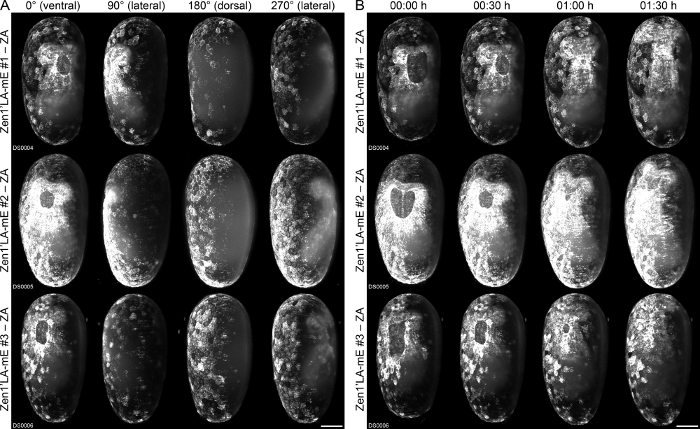
Figure 5: Comparative fluorescence live imaging of one embryo each from the Zen1’LA-mE #1, #2 and #3 transgenic Tribolium sublines. (A) Embryos are shown along four directions during gastrulation, just prior to serosa window closure. (B) Embryos are shown ventrally over a period of 01:30 h (from a total imaging period of 118:00 h). Image adjustment was performed with identical minimum and maximum displayed values, no dynamic intensity correction over time was performed. Metadata for the Tribolium datasets can be found in Supplementary Table 1. Datasets were synchronized to the stage shown in (A). ZA, Z maximum projection with image adjustment. Scale bar, 100 µm. Please click here to view a larger version of this figure.
| Combination | Rationale | Illumination lens | Detection lens | Camera | Reference |
| #1 | entire embryo on the cellular level, large pixel spacing (small data volume) | 2.5× magnification numerical aperture 0.06 (air) | 10× magnification numerical aperture 0.3 (water-dipping) | CCD with 1.4 megapixel (1392×1040, 6.45 µm pitch) | this study, Drosophila41, Tribolium20,41 |
| #2 | entire embryo on the cellular/subcellular level, small pixel spacing (large data volume) | 2.5× magnification numerical aperture 0.06 (air) | 20× magnification numerical aperture 0.5 (water-dipping) | sCMOS with 5.5 megapixel (2560×2160, 6.5 µm pitch) | Tribolium9 |
| #3 | specific regions of the embryo on the subcellular level or specialized applications e.g. live imaging of dissected germbands42,43 | 5.0× magnification numerical aperture 0.16 (air) | 40× magnification numerical aperture 0.75 (water-dipping) | CCD with 1.4 megapixel (1392×1040, 6.45 µm pitch) | Tribolium44 |
Table 1: Recommended illumination lens/detection lens/camera combinations for live imaging of Drosophila and Tribolium embryos using LSFM. All combinations have been successfully employed in previous studies (see Reference row). Please note that most sample chamber-based LSFMs use water-dipping lenses for detection, which are primarily designed for imaging buffers with a refractive index of 1.33 (such as autoclaved tap water or other aqueous media). Imaging buffers with other refractive indices can be used40, but this changes certain properties of the optical system, e.g. the working distance.
| Line | Genotype | Fluorophore expression |
| #01691 | y[1] w[67c23]; P{w[+mC]=Ubi-GFP.nls}ID-2; P{Ubi-GFP.nls}ID-3 | NLS-GFP under control of the polyubiquitin promoter |
| #29724 | w[*]; P{w[+mC]=Tub84B-EGFP.NLS}3 | NLS-GFP under control of the alpha-tubulin 84B promoter |
| #24163 | w[*]; P{w[+mC]=His2Av-EGFP.C}2/SM6a | EGFP-labeled histone 2A variant in all cells |
Table 2: The three transgenic Drosophila lines used in the first application example. The “line” column refers to the Bloomington Drosophila Stock Center (bdsc.indiana.edu) stock number.
Supplementary Video 1: Comparative fluorescence live imaging of one embryo each from the #01691, #29724 and #24163 transgenic Drosophila lines. Each transgenic line expresses nuclear-localized EGFP/GFP under control of a presumably ubiquitous and constitutively active promoter. ZA, Z maximum projection with image adjustment. No dynamic intensity correction over time was performed. ZA, Z maximum projection with image adjustment. Please click here to download this video.
Supplementary Video 2: Comparative fluorescence live imaging of one embryo each from the Zen1’LA-mE #1, #2 and #3 transgenic Tribolium sublines. Each transgenic subline expresses mEmerald-labeled Lifeact under control of the zerknüllt 1 promoter, a transcription factor involved in serosa specification. Please note that the Zen1’LA-mE #1 embryo turns approximately 90° within the serosa right after serosa window closure. Datasets were synchronized to the stage shown in Figure 4A. Image adjustment was performed with identical minimum and maximum displayed values, no dynamic intensity correction over time was performed. Embryos are shown ventrally and laterally over a period of 48:00 h. ZA, Z maximum projection with image adjustment. Please click here to download this video.
Supplementary File 1: FIJI batch processing script for the automated calculation of z maximum projections from all z stacks within one folder. The script can be opened in FIJI via drag-and-drop. At the start (“Run”) only the input folder must be specified. The output subfolder ("Z Maximum Projections") is created automatically. Please click here to download this file.
Supplementary Table 1: Metadata and parameter for the long-term live imaging datasets DS0001-6. Please click here to download this table.
