Romtemperatur (RT) makromolekylær krystallografi er populært igjen innen strukturbiologi samfunnet. Utviklingen av X-ray Free Electron Laser (XFEL) lyskilder har ansporet utviklingen av RT-prøveleveringsmetoder 1,2,3,4, og disse metodene har nå blitt brukt på synkrotroner 5,6,7,8. Ikke bare åpner RT-metoder muligheten for pumpesondeeksperimentelle strageties 9,10,11,12, men det er også økende bevis på at de fremmer alternative konformasjonstilstander innen proteiner 13,14,15,16,17.
Imidlertid var hovedårsaken til at kryo-metoder fikk trekkraft over RT-tilnærminger på slutten av 1990-tallet bremset strålingsskader ved krystalltemperaturer undernull 18. Kryo-metoder19 begynte å tillate innsamling av et komplett datasett fra en enkelt proteinkrystall. Moderne RT-metoder ved XFEL og synkrotroner løste problemet med enkeltkrystallstrålingsskader ved utvikling av raske (> 100 Hz) krystallleveringsstrategier 1,2,3,4. Disse metodene tillater innsamling av et komplett datasett fra tusenvis av individuelt eksponerte krystaller. Disse RT-leveringsmetodene krever derfor produksjon av store mengder løsninger som inneholder homogene mikrokrystaller (> 100 μL < 50 μm krystaller). Men siden kryo-metoder har en tendens til å bare kreve enkeltkrystaller, er metoder for å lage slike mikrokrystallinske oppslemminger for tiden ikke allestedsnærværende på tvers av proteinkrystallografilaboratorier.
Det finnes eksempler i litteraturen på hvordan man utfører deler av mikrokrystallisasjonsoptimaliseringsprosedyren for seriekrystallografiprøver. Her bør det skilles mellom membran og løselige proteiner. Protokoller for å optimalisere veksten av mikromembranproteinkrystaller dyrket i monoolein (eller et annet lipid), for lipidisk kubisk fase (LCP), har blitt godt beskrevet20,21,22. Imidlertid mangler metoder for mikrokrystallisering av løselige proteiner, inkludert membranproteiner dyrket under ikke-LCP-forhold, generelt. Tidligere studier har fokusert på spesifikke deler av prosessen, for eksempel mikrokrystallscreening 23,24, forbedring av nukleering24 og skalering ved bruk av fri grensesnittdiffusjon 25, men ikke en komplett metode.
Imidlertid ble det nylig beskrevet en metode26 som forsøker å tilby en komplett protokoll. Som mange aspekter av proteinkrystallografi er det ikke nytt. Mange av de foreslåtte ideene ble allerede beskrevet av Rayment (2002) 27. Metoden tar sikte på å vise krystallografer hvordan man utfører konverteringen fra en enkelt krystall dyrket ved hjelp av dampdiffusjon, til en batchmetodikk for å dyrke tusenvis av mikrokrystaller. Metoden fokuserer på dampdiffusjon som et vanlig utgangspunkt, da 95% av alle Protein Data Bank (PDB) avsetninger kommer fra krystaller dyrket i dampdiffusjonsplater26. Dampdiffusjon er imidlertid ikke den ideelle metoden for mikrokrystallisering26, så en metodikk er beskrevet for å konvertere dampdiffusjon til batchkrystallisering. Når krystaller kan dyrkes i batch, blir skaleringsruter til større volumer mer praktisk. Gitt vagaries av proteinkrystallisering, vil forfatterne understreke at denne metoden ikke er feilsikker. Imidlertid bør protokollen i det minste gi et innblikk i “krystallisasjonsrommet” til et protein.
Denne metoden er avhengig av proteinkrystallisasjonsfasediagrammet og hvordan en forståelse av diagrammet kan fungere som en veiledning under mikrokrystallisasjonsoptimalisering. Et proteinfasediagram er ofte avbildet som et x/y-plott med utfellings- og proteinkonsentrasjoner på henholdsvis x– og y-aksen (figur 1A). Fra rentvannspunktet (nederst i venstre hjørne – figur 1A) øker konsentrasjonen av både protein og utfelling til løselighetslinjen er nådd. Løselighetslinjen markerer overmetningspunktet (lilla linje – figur 1A). Når et protein er overmettet, blir løsningen termodynamisk ustabil og vil begynne å separere seg i to faser: “proteinrik” og en stabil mettet løsning. Denne separasjonen kan forekomme hvor som helst utover løselighetslinjen, og dens kinetikk er avhengig av egenskapene til proteinet og komponentene i løsningen.
Når protein- og utfellingskonsentrasjonene er for store, vil proteinet brytes ustabilt ut av løsningen og resultere i amorft bunnfall (rosa region – figur 1A). Imidlertid kan bestilt faseseparasjon forekomme i kjerneområdet [se Garcia-Ruiz (2003) 28 for detaljert beskrivelse] og krystallkjerner har tilbøyelighet til å danne (grønn region – figur 1A). Nukleering og vekst fjerner protein fra løsningen og beveger dråpen inn i den metastabile regionen hvor veksten kan fortsette til løselighetslinjen er nådd [se McPherson and Kuznetsov (2014) 29 for detaljert diskusjon . Diagrammet er for de aller fleste krystallisasjonsforhold en grov overforenkling30. Uavhengig av dette er diagrammet fortsatt til stor nytte for mikrokrystallografer, da kartleggingen av diagrammet gjør det mulig å bestemme løselighetslinjen og kinetikken for kjernedannelse.
Når det gjelder å lage mikrokrystaller, er de to faktorene under krystallisering som må optimaliseres, antall krystaller (Xn) og deres gjennomsnittlige, lengste dimensjon (Xs). X n vil være proporsjonal med antall nukleasjonshendelser (n ) (Eq. 1).
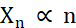 Ekv. 1
Ekv. 1
X s er proporsjonal med konsentrasjonen av fritt protein overløselighetslinjen (Ps) delt på Xn (Eq. 2).
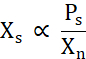 Tilsvarende 2
Tilsvarende 2
I en perfekt situasjon ville hver nukleasjonshendelse gi en mulig krystall, og hver og en av disse krystallene ville ha lik tilgang til det tilgjengelige proteinet i oppløsning. Figur 2 er en grafisk fremstilling fra et ideelt scenario av forholdet mellom Xn og Xs. I praksis er hovedkontrollen en krystallograf har over X,n og Xs ved å påvirke mengden kjernedannelse eller ved tilsetning av frøkrystaller. Mikrokrystallografen må bedømme hvordan man kan øke Xn slik at en passende krystallkonsentrasjon og krystallstørrelse begge kan opprettes.
De fleste krystallisasjonsteknikker krever en “overgangsperiode” (figur 1B). For eksempel, i et dampdiffusjonseksperiment, ved blanding av protein- og utfellingsløsninger, vil konsentrasjonene av hver endre seg når dråpen likevekter med brønnløsningen. Man håper at disse endringene gradvis vil overføre dråpen til nukleasjonssonen hvor tilbøyeligheten til krystallisering vil øke. Når krystaller begynner å kjerne og vokse, vil mengden protein i oppløsning begynne å falle, noe som reduserer sannsynligheten for ytterligere nukleering. Den endelige mengden kjernedannelse vil være protein- og tilstandsspesifikk, og også avhengig av dybden av penetrasjon av inn i nukleasjonssonen. Gitt den begrensede nukleasjonssonepenetrasjonen av metoder som krever et overgangstrinn, vil nukleasjonsnivået til slutt være begrenset til nukleasjonshastigheten ved grensen for metastabil-nukleasjonsområdet.
På grunn av viktigheten av å kunne øke nivået av nukleering for en mikrokrystallograf, er det viktig å flytte til en batchkrystallisasjonsmetodikk. Batch kan dra større nytte av hele nukleasjonsområdet (figur 1C). I batchmetoder er ideen å blande protein og utfelling sammen slik at en overmettet løsning opprettes uten behov for endringer i komponentkonsentrasjoner. Nukleering bør være mulig umiddelbart etter blanding. Batchmetoder tillater derfor at hele nukleeringssonen teoretisk nås. Enhver økning i nukleasjonskinetikken utover den metastabile nukleasjonsgrensen kan deretter utnyttes.
Hvis basalnivået av krystallkjernedannelse ikke er nok til å generere en stor Xn, kan mikrosåingsmetoder brukes. I mikrosåing brytes forvokste krystaller opp for å lage en oppslemming av krystallinske fragmenter som kan fungere som et stillas for frisk krystallvekst31,32. Mikrosåing har blitt mye brukt i seriell krystallografisk prøvepreparering som en måte å øke Xn uten behov for økende krystallkjernedannelse (figur 1C).
Overgangen fra dampdiffusjon til batch kan visualiseres på et fasediagram som å flytte det eksperimentelle utgangspunktet fra enten de ikke-overmettede eller metastabile områdene til nukleeringssonen. Dette kan gjøres ved å øke protein- og/eller utfellingskonsentrasjonene, og/eller forholdet mellom de to i dråpen (figur 1D), og observere hvilke forhold som gir krystaller som opptrer raskt (< 24 timer)26. Fullstendig dampdiffusjonsdråpebalanse kan ta dager eller uker33. Derfor, ved å lete etter forhold som viser raskt forekommende krystaller, kan batchforhold bli funnet uten å måtte flytte til alternative krystallisasjonsscreeningsformater som mikrobatch34,35,36,37.
Når nukleasjonssonen er funnet, er det funnet en batchtilstand og et morfogram – her et grovfasediagram – kan opprettes. Morfogrammet er til stor nytte når du vurderer om du skal bruke en sådd batch eller rett batchprotokoll. Ved å plotte Xn som funksjon av protein- og utfellingskonsentrasjonen, kan det gjøres en vurdering av nukleasjonskinetikken26. Hvis X n forblir lav over hele kjerneområdet, kan det være nødvendig med frøbatch for å gjøre Xn stor nok til å begrense krystallveksten. Denne vurderingen er det første trinnet i prosessen med å skalere til større volumer (> 100 μL).
Denne metoden ble utformet slik at den kunne utføres i de fleste krystallisasjonslaboratorier ved bruk av standard dampdiffusjonskrystallisasjonsutstyr. Det er også gjennomført mange studier som beskriver teknikker for å legge til rette for mange deler av denne prosessen, dersom utstyret skulle være tilgjengelig. Disse inkluderer, men er ikke begrenset til, dynamisk lysspredning (DLS) 25,27, ikke-lineær avbildning 20,24,25, pulverdiffraksjon 20,24,27 og elektronmikroskopi 26 [se Cheng et al. (2020) 40 for en fin gjennomgang].
Målet med dette arbeidet er å gi en visuell demonstrasjon av metoden for overgang fra lite volum ( 100 μL) batchkrystallisering. Endothiapepsin fra Cryphonectria parasitica har blitt brukt som et eksempelsystem for å demonstrere denne oversettelsen. Typen eksperiment og prøveleveringsmetode som mikrokrystallene er nødvendige for, vil påvirke den ideelleXs utgang26. For blandingsforsøk som krever en millisekunds tidsoppløsning41 eller gassdynamiske virtuelle dyser42, kan en endelig Xs på < 5 μm være ønskelig. I dette tilfellet var målet å produsere proteinkrystaller som diffrakterer til ca. 1, 5 Å, for et fotonaktivert pumpesondeeksperiment, og ved hjelp av en fast målleveringsmetode.
For å gi en illustrasjon av prøvekravene til et slikt seriekrystallografieksperiment ved bruk av endothiapepsin, viser tabell 1 de eksperimentelle parametrene til et hypotetisk eksperiment. Eksempelinformasjonen var basert på protokollen beskrevet nedenfor. Gitt noen konservative estimater på trefffrekvenser og datainnsamlingskrav, er 50 mg det totale prøveforbruksestimatet for hele eksperimentet.
Figur 3 viser et flytskjema over hele optimaliseringsprosessen fra første lille volum dampdiffusjonskrystallisering til storskala batch. For de fleste seriekrystallografiprosjekter vil denne protokollen begynne på trinn 2: “overgang til batch”, siden målproteinet allerede vil ha blitt krystallisert. Trinn 1 er imidlertid inkludert for fullstendighet og for å minne leserne om dens betydning. Å finne en tilstand som gir opphav til en brønndiffracting, enkelt, stor krystall er det beste utgangspunktet for mikrokrystalloptimalisering. I trinn 2 kan denne tilstanden deretter optimaliseres fra dampdiffusjon til batch, og et morfogram av nukleerings- og metastabile regioner kan plottes. Når dette er gjort, kan skalering av batchbetingelsen til større volumer utføres i trinn 3. Ved slutten av flytskjemaet vil en krystallograf ha opprettet en repeterbar, storvolum (> 100 μL), mikrokrystallisering, batchprotokoll for endothiapepsin. Denne metoden kan deretter brukes på deres spesielle protein av interesse.
