To avoid mistargeting, before every experiment, verify the coordinates using dye injections. Animals were injected with 0.2-0.5 µL tryptophan blue using the same protocol, capillary was rapidly withdrawn after injection and the brain was quickly frozen to avoid diffusion. After sectioning on the microtome, the injection site can be seen in blue (Figure 2 C,E). To ensure effective targeting, dye injections should be carried out successfully on 2-3 animals prior to actual experiment.
Stereotaxic injections can be used to create three main Parkinson's disease models with varying degrees of pathology and neurodegeneration. Typically, in the PD field, neurodegeneration is evaluated by quantifying tyrosine hydroxylase (TH), a marker for dopaminergic neurons, as well as a general neuronal marker (e.g., Neuronal Nuclei [NeuN] or HuC) as downregulation of TH due to a toxic environment can give the illusion of nigral cell death.
When injecting toxicants such as 6-OHDA the goal is to eradicate as many nigral dopaminergic cells as possible. 6-OHDA is taken up by monoamine transporters and blocks mitochondrial respiration21. Desipramine is given before each surgery to ensure the toxicant is only taken up by dopamine neurons and not by noradrenergic or serotonergic cells. To evaluate the success of each injection, fixed midbrain sections should be Immunolabeled for TH and NeuN. As toxins act quickly, this model only takes 2-3 weeks after injection to be fully developed. If 6-OHDA injections into the medial forebrain bundle (MFB) were successful, dopaminergic cell loss should be 80% and greater compared to PBS injected controls (Figure 2 F-G). The MFB is a region where nigrostriatal projections bundle together and therefore allow for targeting many dopaminergic neurons with a single injection. Toxicants can also be injected directly into the nigra or striatum for less severe cell death.
Viral vector-based models highly depend on the (sero-)type of the vector used. AAV2 was the most commonly injected as it was easy to produce and has a high affinity towards dopaminergic neurons. Engineering of expression promoters, viral capsids and envelope proteins has opened doors to more specific targeting of brain regions and cell populations. Most viral vectors in PD models are directly injected into the SN to transduce dopaminergic neurons in the pars compacta. Expression of most vectors stabilizes 3 weeks after injection and can be visualized by immunolabeling for the exogenous protein. Usually, GFP is expressed as a control at titers not causing neurodegeneration (Figure 2H). PD-related genes are used to mimic the genetic aspect of the disease, for example duplications or triplications of asyn. Overexpression of asyn in nigral neurons causes dopaminergic cell death (Figure 2I).
The PFF model is the newest addition to the PD tool kit. Aggregated asyn can be acquired either in vitro by weeklong shaking of recombinant asyn protein to generate PFFs or by isolating aggregates from animal models or patient brains. Injected PFFs or brain extracts are then causing accumulation in neurons projecting to the injection site. This model is unique in that it relies on triggering normal endogenous asyn to phosphorylate and accumulate rather than driving aggregation via elevating endogenous asyn. Asyn PFFs are commonly injected into the striatum. Several weeks following injections, asyn pathology can be visualized in prefrontal cortex, striatum, cortex and SN by immunolabeling for phosphorylated asyn (Figure 2K). Control injections of PBS or serum albumin do not result in those PD typical intraneuronal accumulation (Figure 2J).
The stereotaxic injection method can be challenging and expensive, and common errors can usually only be detected after the animals have been sacrificed. The capillary thickness of the blunt end can vary from 10-100 µm depending on the pulling method. If the capillary is too thin, it can clog while entering the brain and lead to a failure in injection (Figure 2L). In this particular case, the needle tract can be identified by scar tissue and uncleared blood cells while no effect of the injection is visible. Another common error is mistargeting (Figure 2M). With age, bregma can be difficult to determine, incorrect coordinates or reading errors can all lead to incorrectly placed injections. While targeting is less complicated in bigger animals (e.g., rats) and larger brain areas (e.g., striatum), they become increasingly more challenging in mice SN injections for example. Another common mistake is dosage. Control substances should not cause more than 10-15% cell death on their own. If expression of GFP (Figure 2N) or PBS injection is toxic, parameters need to be changed to ensure that pathology and cell death are specific to the substance injected.
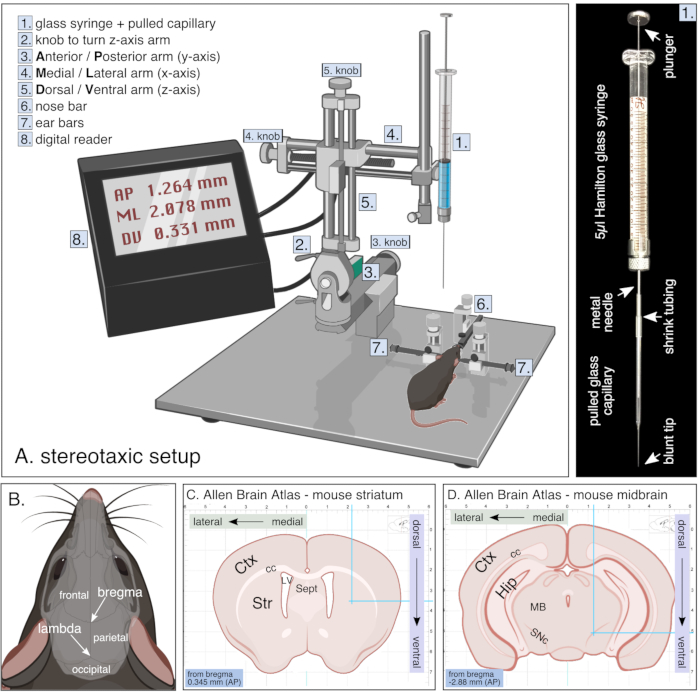
Figure 1. Setup for stereotaxic surgery. (A) Schematic of a stereotaxic setup during survival surgery including digital reader and glass syringe which is further detailed on the right. (B) Skull anatomy of a rodent showing bone plates (frontal, parietal and occipital bones) with corresponding sutures resulting in bregma and lambda. (C,D) Adapted images from the Allen brain atlas webpage showing coronal sections of mouse striatum (C) and midbrain (D) with corresponding coordinate grid in mm. Images adapted from biorender. Please click here to view a larger version of this figure.
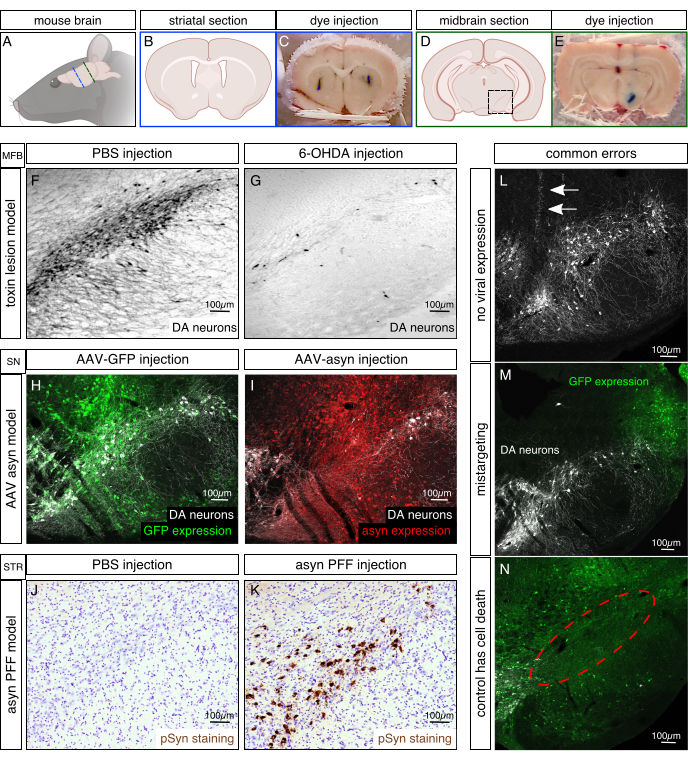
Figure 2. Histological examples of Parkinson's disease animal models. (A) Schematic of the orientation of a mouse brain. Blue line indicates location of coronal striatal section for B and C. Green line indicates location of coronal midbrain section in D and E. (B,D) Illustrations of coronal mouse brain section showing the striatal area in B and the midbrain area in D. Black box represents magnified ventral midbrain (substantia nigra) region in F-N. (C,E) Coordinates were tested for their accuracy prior to experiment by injecting 0.2 µL tryptophan blue. Brain was immediately taken out after surgery, frozen and cut on a microtome to analyze targeting. Examples for correct targeting of striatum and substantia nigra are shown in C and E, respectively. (F,G) Representative images of the 6-hydroxydopamine (6-OHDA) model are shown by immunolabeling for the dopaminergic marker tyrosine hydroxylase (TH) on nigral sections. 6-OHDA (4 µL of 3 µg/µL; G) or PBS as control (F) was injected into the medial forebrain bundle (MFB) and rats were sacrificed 6 weeks after surgery. Half an hour prior to injection, 25 mg/kg desipramine was given i.p. to prevent noradrenergic cell death. (H,I) Example of viral vector based animal model shows transduced cells in green (Immunolabeled against GFP transgene [control vector], H) or red (Immunolabeled for asyn, I), and DA neurons in white. AAVs with a serotype 6 (1 µL of 7 x 1013 viral genomes /mL each) were injected directly into the substantia nigra pars compacta (SN) and mice were sacrificed 4 weeks after surgery. (J,K) Representative images of rats injected with 8 µg of mouse asyn PFF (K) or with PBS as a control (J) were Immunolabeled for phosphorylated asyn (brown) and cell nuclei with cresyl violet (purple). Rats were injected into the striatum (2 x 2 µL deposits) and sacrificed 8 weeks after injection. Images adapted from Duffy et al.21. (L,M,N) Common errors of stereotaxic experiments are no viral solution injected (L, needle tract indicated by arrows), mistargeting (M, in this case too lateral) and control injection causes cell death (N, missing cells indicated by red circle). Transduced neurons are Immunolabeled in green for GFP and white for TH. Please click here to view a larger version of this figure.
