Several representative datasets have been published In the past few years10,12 along with the crystallographic results and scientific findings from a diverse range of protein samples, including photoreceptor proteins and enzymes, for example, a plant UV-B photoreceptor UVR8, a light-driven DNA repair photolyase PhrB10, a novel far-red-light sensing protein from a multi-domain sensory histidine kinase14, ligand/light dual-sensor domains, and the photosensory core module of a bacteriophytochrome12. As representative results, we list the on-chip crystallization conditions of these proteins in Table 1, and directly compare them with the conditions used for the vapor diffusion method. Here we show four additional case studies of on-chip crystallization (Figure 2) and a collection of in situ diffraction patterns in a movie (Movie 2). Representative in situ datasets collected using this protocol are summarized in Table 2.
In a representative case, cryocrystallography gave rise to poor diffraction for a far-red-light sensing photoreceptor protein likely due to light sensitivity and high solvent content (~80%) of these crystals14. The electron densities obtained from the cryocrystallography data were too smeared to resolve the chromophore conformation, which is at the center of our scientific question. Using the in situ protocol, we were able to avoid unintended light activation before diffraction and obtained a dark dataset at room temperature from more than 800 crystals. This dark dataset from in situ serial Laue diffraction resulted in better resolved electron densities, allowing confident model building of a bilin chromophore that exhibits a hitherto unknown all-Z,syn conformation (Figure 7A)12,14. Our dynamic crystallography experiments have further revealed light-induced changes in this far-red photoreceptor protein by comparing data from 4,352 crystals in dark and 8,287 crystals after light illumination (Figure 7). A preliminary analysis of the light-induced difference maps has revealed concerted motions in the central β sheet, suggesting the importance of the π-π stacking between the pyrrole rings of the chromophore and several aromatic residues (Figure 7B,C). An in-depth analysis and scientific findings will be presented elsewhere.

Figure 1: Crystallization device assembly. Each assembly is estimated to cost US$30 with two monocrystalline quartz wafers or US$10 with two glass coverslips. Hardware components except the shim are reusable. (A) The flat side of the outer ring is labeled for identification purposes. (B) The outer ring is placed upside down on a clean surface. (C) A quartz wafer of 1 inch in diameter is carefully placed inside. A glass chip could also be used instead during crystallization trials but is not compatible with X-ray diffraction. (D) Both sides of the shim are oiled. (E) The oiled shim is placed on the first quartz chip. (F) Protein and crystallization solutions are pipetted to the center of the chip and mixed. (G) A second quartz or glass chip covers the drop so that it evenly spreads over the chip. (H) A retaining ring is screwed over the second quartz wafer. (I) A tightening tool is used to tighten the retainer ring gently. (J) A fully assembled device. Please click here to view a larger version of this figure.
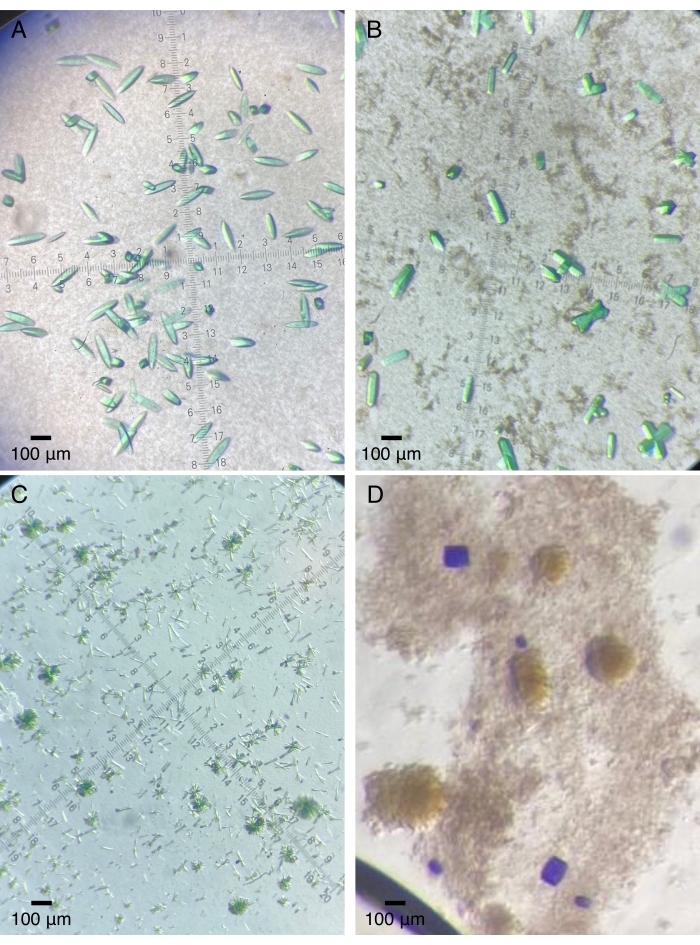
Figure 2: Representative protein crystals grown on quartz devices. (A) The photosensory core module of a bacteriophytochrome (Pa497 in Table 1). (B,C) Different constructs of the third GAF domain from a multi-domain sensory histidine kinase (2551g3 and 2551g3Δα1 in Table 1). (D) The tandem sensory domains from a dual-sensor histidine kinase (RECGAF in Table 1). Please click here to view a larger version of this figure.

Figure 3: inSituX diffractometer. (A) A crystallization device is mounted in the chip holder. Although the device is mounted vertically, crystals grown on chip are not going to fall, primarily because the liquid layer in an assembled device is very thin, and crystals are anchored to their nuclei when growing. (B) An IR light source is installed for the optical scan. The camera captures the inline view of the protein crystals along the X-ray beam through a prism mirror (not visible in the picture). Please click here to view a larger version of this figure.
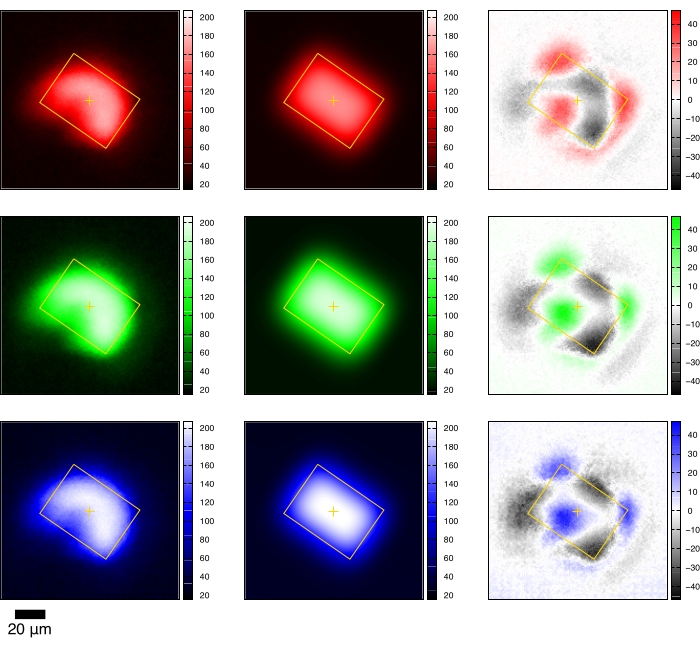
Figure 4: Direct beam profile fitting. The red, green, and blue channels of the X-ray fluorescence image are used to fit a two-dimensional Gaussian function. The left column shows the raw image of the red, green, and blue channels. The middle column is the fitting result with the precise beam position and size. The right column displays the fitting residuals. If the amplitude of the fitting residuals spans a small fraction of the raw image, the profile fitting of the direct beam is successful. Please click here to view a larger version of this figure.
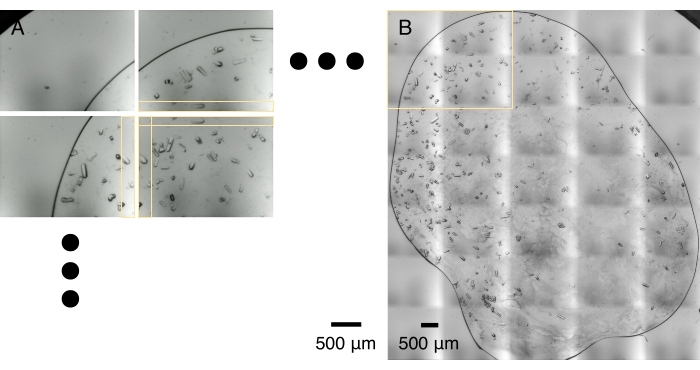
Figure 5: Image tiling. (A) An array of crystal micrographs is captured during an optical scan. The optical scan and data transfer usually take 1-2 min. Adjacent micrographs share a strip of overlapping area, horizontally and vertically, as marked by yellow boxes. (B) Micrographs are stitched together to make a high-resolution montage based on the optimal correlation in the overlapping areas. This process usually takes a minute on a laptop computer. The yellow box outlines the area captured by 2 x 2 micrographs shown in (A). Please click here to view a larger version of this figure.
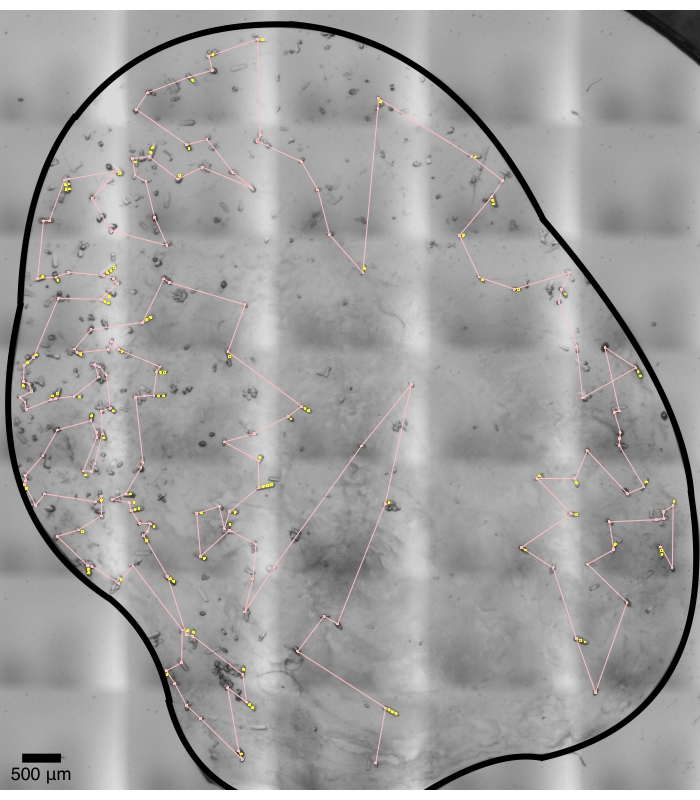
Figure 6: Crystal recognition and shot planning. Each pink circle marks the primary shot of a crystal. The yellow circles mark additional shots if a crystal is long enough to position these shots. The pink lines mark a route as a solution to the traveling salesman problem. Clustered crystals and smaller crystals are largely avoided. The aggressiveness of crystal finding can be adjusted as an option to findX.py (step 5.6). A brute force "overkill" strategy would leave no crystal un-shot but could produce lots of diffraction images, but not processable12. Please click here to view a larger version of this figure.
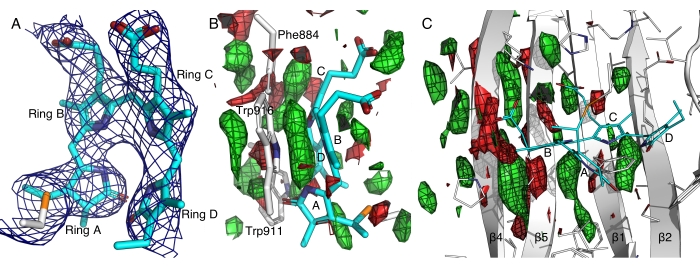
Figure 7: Electron density maps of the far-red-light sensing domain of a histidine kinase. (A) The 2Fo-Fc map contoured at 2.5σ shows the electron densities associated with the bilin chromophore in an all-Z,syn conformation14. Pyrrole rings A through D are marked. (B and C) Light-dark difference maps contoured at ±2.5σ in green and red, respectively, highlight the gain and loss of electron densities. Please click here to view a larger version of this figure.
Table 1: Comparison of crystallization conditions between vapor diffusion and on-chip batch method. Vapor diffusion and batch methods of crystallization are highly correlated10,14,15,16,17,18,19. Starting from a vapor diffusion condition, a similar condition can be optimized for on-chip crystallization. Please click here to download this Table.
Table 2: Summary of in situ datasets collected directly from quartz devices. Thousands of Laue diffraction patterns can be collected from several crystallization devices. Please click here to download this Table.
Movie 1: A mock data collection. Targeted crystals are translocated into the X-ray beam as marked by a red circle. The sequence of the targeted crystals in this movie does not follow a solution to the traveling salesman problem. Laser and X-ray exposures are fired at each stop with a programmed delay. Diffraction images are collected. Please click here to download this Movie.
Movie 2: Diffraction images. Hundreds of diffraction images can be collected from a single crystallization device. Several devices are sufficient to produce a complete and highly redundant dataset (Table 2). Please click here to download this Movie.
Supplementary File 1: Sample <device>.param file. A small text file collects some control parameters specific to each crystallization device. These parameters start with their default values and will be modified accordingly in sections 4, 5, and 6 as the protocol proceeds. Please click here to download this File.
