In this protocol, HepG2/C3A spheroids were treated with 20 mM NaBut and 10 mM NaSuc, both of which affected global levels of histone PTMs (Figure 3A). Histone PTMs were then identified and quantified at the single residue level via MS/MS acquisition (Figure 3B).
When samples are run in replicates, statistical analysis can be performed to assess the fold change enrichment of a PTM between samples, as well as the reproducibility of the observation. The data shown demonstrate that peptides modified with acetylations are enriched in spheroids treated with NaBut vs. control (Figure 3C), while samples treated with NaSuc have a higher relative abundance of histone peptides modified with lysine succinylation (Figure 3D). These calculations were done in a spreadsheet program as detailed in a separate publication34. An overall increase of a given histone modification can be better represented in radar plots, where observing a higher global abundance of a certain modification becomes more intuitive, even while maintaining detailed information about the modification sites analyzed (Figure 3E,F).
This protocol generates a peptide from histone H3 amino acid residues 9-17, which include the frequently modified residues K9, S10, and K14. The data shown indicate that treatment with NaBut increases the levels of H3K14ac, but only on histones co-modified with H3K9me2 and not H3K9me3 (Figure 4A). The co-existence frequency between two modifications can be represented more intuitively as a ring graph, where the nodes represent individual modifications while the thickness of connector lines represents the co-existence frequency between the two PTMs (Figure 4B). Sometimes, the co-existence frequency is unaffected, but data represented as bar plots might be misleading. For instance, the data represented in Figure 4A indicate that the combination H3K9me2K14ac is more abundant in NaBut treatment than in control. This is correct, but this given combination is the most frequent regardless of the treatment. Figure 4B clearly shows that H3K9me2K14ac and H3K9me3K14ac are the most frequent combinatorial patterns regardless of the treatment (line thickness), but that global levels of H3K14ac (node) are what is truly changing in the experiment.
This protocol generates a peptide from histone H4 residues 4-17, which includes modifiable residues at positions K5, K8, K12, and K16 (mostly by acetylations). When comparing control and NaBut treatment, it is possible to observe an increase in combinations of acetylations by representing data as, for example, word clouds (Figure 4C). This representation clearly highlights that the unmodified version of histone H4 is most abundant in the control sample, while spheroids treated with NaBut are enriched in doubly, triply, and quadruply acetylated histone H4 proteoforms. However, word clouds are limited in displaying exact values; the relative abundance of a histone code should be de-convoluted by the size of the text, which may be inaccurately estimated. Therefore, Venn diagrams or more modern equivalents such as UpSetR representation35 can be used to show the exact quantification of co-existing histone PTMs (Figure 4D,E). The data shown highlight once again that selected combinations of acetylations on histone H4 are relatively more abundant in NaBut treatment compared to control.
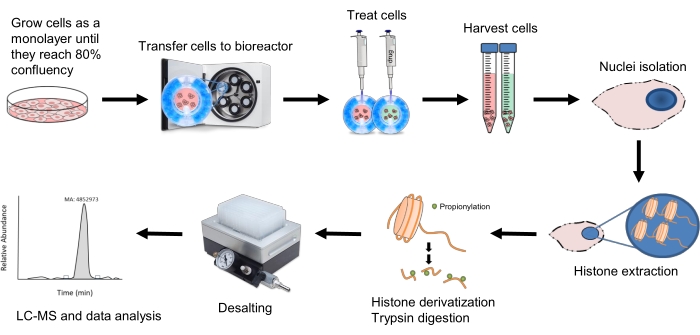
Figure 1: Workflow for histone peptide analysis of 3D spheroids. HepG2/C3A cells are first grown in 2D culture until they reach 80% confluency. The cells are then transferred to an equilibrated bioreactor and placed within the clinostat incubator where they will rotate at 10-11 rpm to form spheroids. After 18 days, the spheroids are treated with either 20 mM NaBut or 10 mM NaSuc and are harvested after their corresponding time points. Nuclei are isolated from the cells and histone extraction is performed with 0.2 M H2SO4. Histone derivatization is then performed with propionic anhydride before and after trypsin digestion to ensure retention of the resulting short peptides by liquid chromatography. Samples are desalted and then run using the LC-MS/MS method mentioned in step 10, and the resulting data is analyzed as described in step 11. Please click here to view a larger version of this figure.
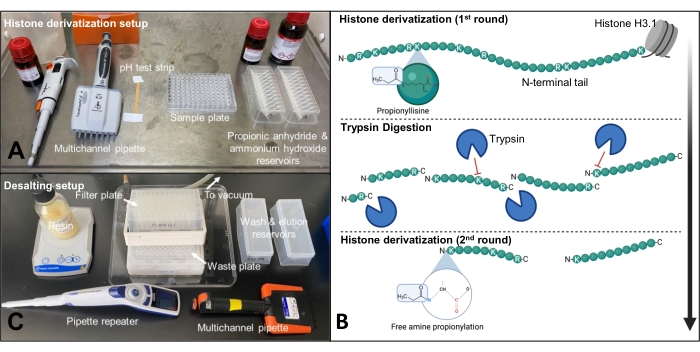
Figure 2: Setup for propionylation and desalting steps. (A) Propionylation is performed in a fume hood and all components are laid out so that the steps can be performed in quick succession. (B) Schematic of first round of propionylation, trypsin digestion, and second round of propionylation on histone H3.1 tail. (C) Desalting is performed on the bench using a 96-well vacuum manifold and a 96-well polypropylene filter plate. Please click here to view a larger version of this figure.
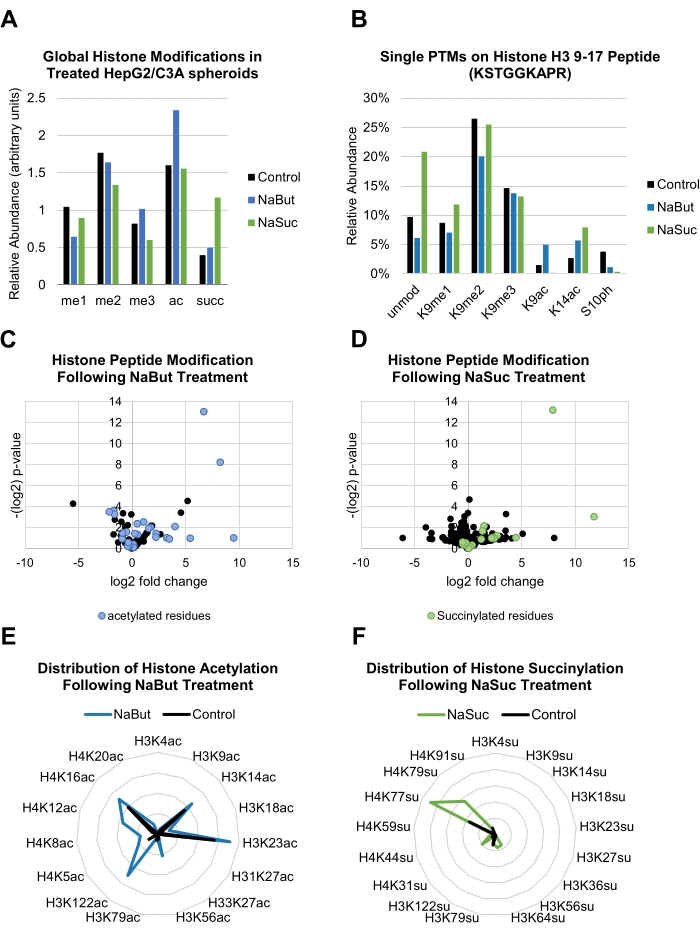
Figure 3: Representation of individual histone modifications. (A) Bar graph showing the relative abundance of common global histone modifications in control and treated (20 mM NaBut or 10 mM NaSuc) HepG2/C3A spheroids. (B) Bar graph showing the abundance of single histone PTMs occurring on residues 9-17 of histone H3 peptide (KSTGGKAPR) in control and treated (20 mM NaBut or 10 mM NaSuc) HepG2/C3A spheroids. (C,D) Volcano plots showing the fold change and significance of differential expression of histone peptide PTMs following treatment with 20 mM NaBut (C) or 10 mM NaSuc (D). Highlighted blue and green points represent acetylated and succinylated residues, respectively. (E,F) Radar plots showing the abundance of single histone peptide acetylation (E) or succinylation (F) following treatment with 20 mM NaBut or 10 mM NaSuc respectively as compared to control. Please click here to view a larger version of this figure.
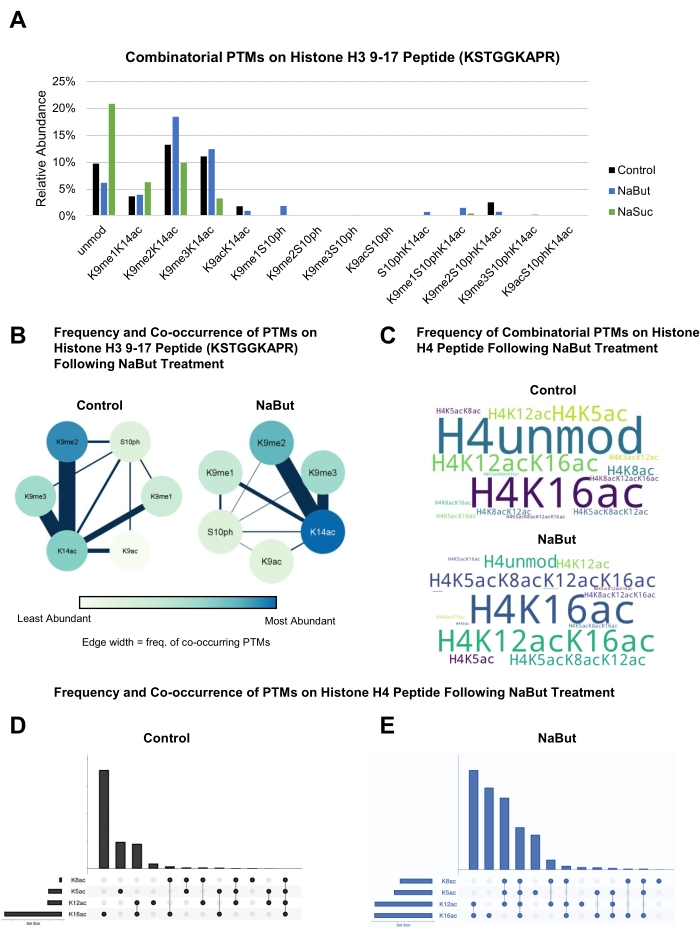
Figure 4: Representation of co-existing histone modifications. (A) Bar graph showing the abundance of combinatorial histone PTMs occurring on residues 9-17 of histone H3 (KSTGGKAPR) in control and treated (20 mM NaBut or 10 mM NaSuc) HepG2/C3A spheroids. (B) Ring graphs showing the relationship between combinatorial histone PTMs on residues 9-17 of histone H3 (KSTGGKAPR) in control and treated (20 mM NaBut) HepG2/C3A spheroids. The intensity of the node color corresponds to the abundance of a single PTM within its treatment group, while the line thickness corresponds to the frequency of PTM co-occurrence. (C) Word clouds showing the frequency of combinatorial histone PTMs on histone H4 residues in control and treated (20 mM NaBut) HepG2/C3A spheroids. The size of the text corresponds with the abundance of the specified combinatorial PTM. (D,E) Venn diagram representing the frequency of co-existing modifications on histone H4 peptide residues 4-17 in control and 20 mM NaBut treated samples. Data are displayed using the ShinyApp UpSetR35. Please click here to view a larger version of this figure.
Supplementary Table 1: List of peptides detected using this protocol. Please click here to download this Table.
