A migration assay was performed on mixed substrates composed of Col1 and high-molecular weight HA (average molecular weight: 1,200-1,400 kDa) using the protocol described here. O9-1 cells at the boundary of the gap were found to readily migrate into the HA-rich gap (Figure 4). Immunostaining for a FA marker, vinculin14, confirmed that the O9-1 cells formed focal adhesions (FAs) at the sites of HA degradation (Figure 5).
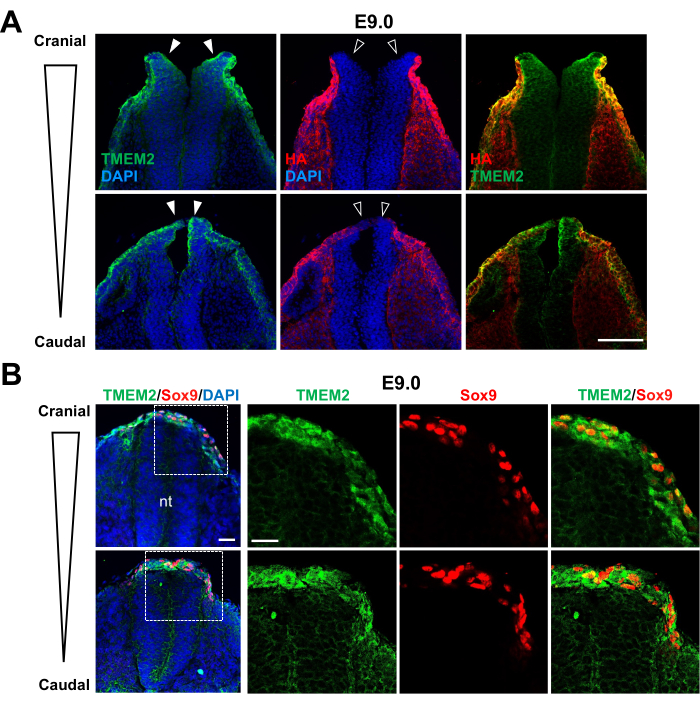
Figure 1: TMEM2 expression in NCCs. Transverse sections of the neural tube of Tmem2-FLAGKI embryos at E9.0. (A) Sections at the cranial and trunk levels of the neural tube were double-labeled for the TMEM2-FLAG protein and HA. TMEM2 expression was observed in the neural plate and the border region of the neural tube (filled arrowheads), whereas these sites were devoid of HA staining (open arrowheads). (B) Double-labeling of the neural crest cells for TMEM2-FLAG and Sox9. Transverse sections of the E9.0 neural tube were stained for TMEM2-FLAG and Sox9. Sox9-positive pre-migratory and emigrating NCCs at the edge of the neural tube highly expressed TMEM2. Abbreviation: nt = neural tube. Scale bars: (A) 300 µm; (B) 100 µm. Adapted with permission from Inubushi et al.9. Please click here to view a larger version of this figure.
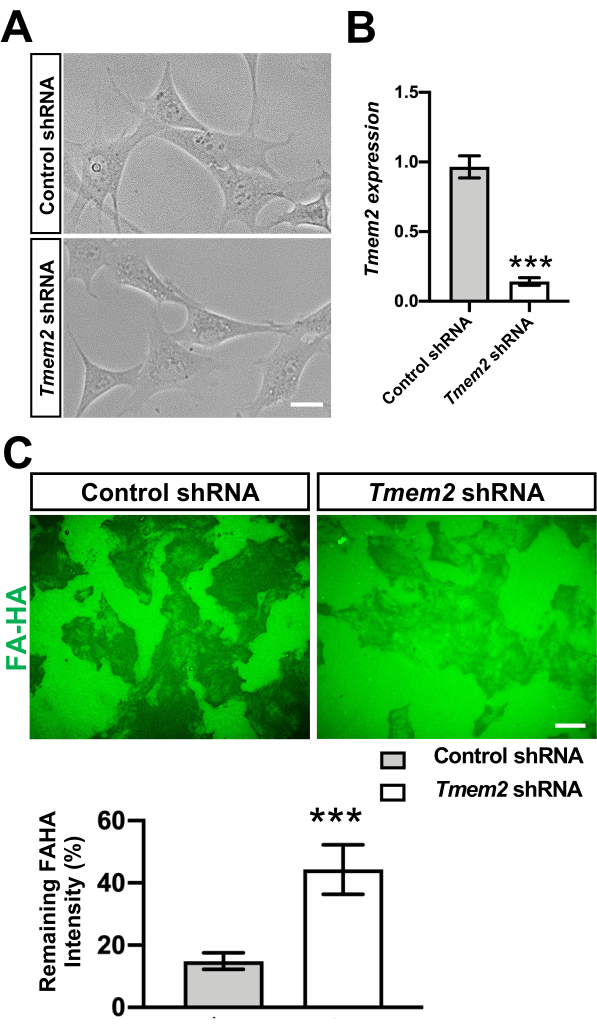
Figure 2: Degradation of HA by TMEM2 in O9-1 cells. (A) Representative images of Tmem2-depleted and control O9-1 cells cultured on a regular culture dish (left). (B) Expression of Tmem2 in these cells was evaluated by qPCR, with Gapdh as an internal control for normalization (bar graph). Means ± SD (n = 5) are shown as horizontal bars. ***p < 0.001 by unpaired Student's t-test. Scale bar, 5.0 µm. (C) Cell-based hyaluronidase assay. Tmem2-depleted and control O9-1 cells were cultured for 48 h on glass coverslips coated with fluoresceinated HA (FA-HA). HA degrading activity is revealed as dark areas in the fluorescent background. The level of HA degradation was also quantitatively compared between Tmem2-depleted and control O9-1 cells as described in Materials and Methods (bargraph). Data represent mean ± SD of the fluorescence intensity underneath a cell relative to that in the cell-free area (n > 50 cells per condition pooled from three independent experiments). ***p < 0.001 by unpaired Student's t-test. Adopted with permission from Inubushi et al.9. Please click here to view a larger version of this figure.
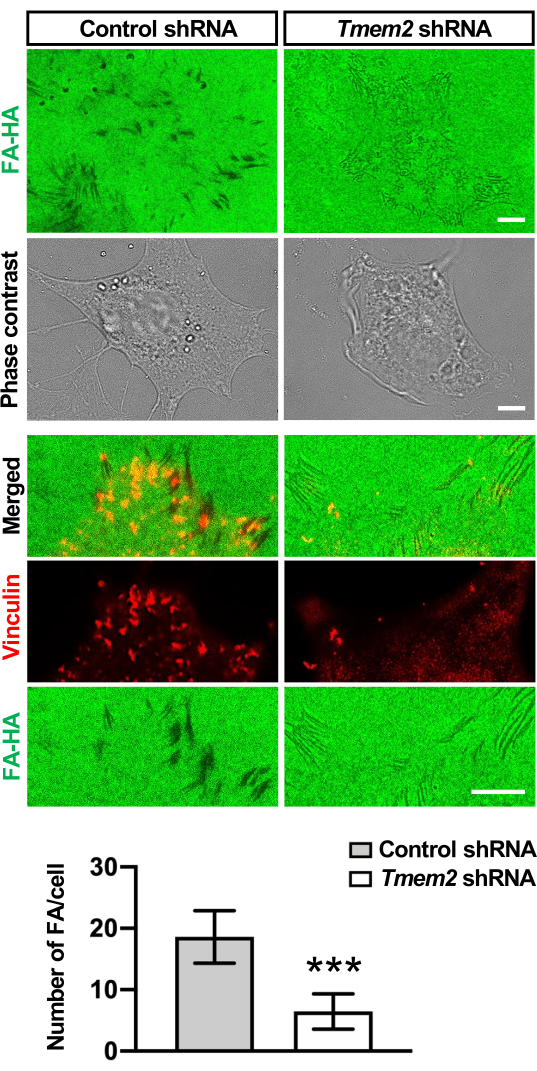
Figure 3: Degradation of substrate-bound HA at FAs in O9-1 cells. Cell-based hyaluronidase assays were performed for 16 h and cells were stained for vinculin. In control O9-1 cells, HA degradation occurs in vinculin-positive FAs. In Tmem2-depleted O9-1 cells, HA degradation and FA formation are greatly diminished. The number of FAs per cell was quantitatively compared between Tmem2-depleted and control O9-1 cells (bar graph). Data represent mean ± SD (n >30 cells per condition pooled from three independent experiments). ***p < 0.001 by unpaired Student's t-test. Adopted with permission from Inubushi et al.9. Please click here to view a larger version of this figure.
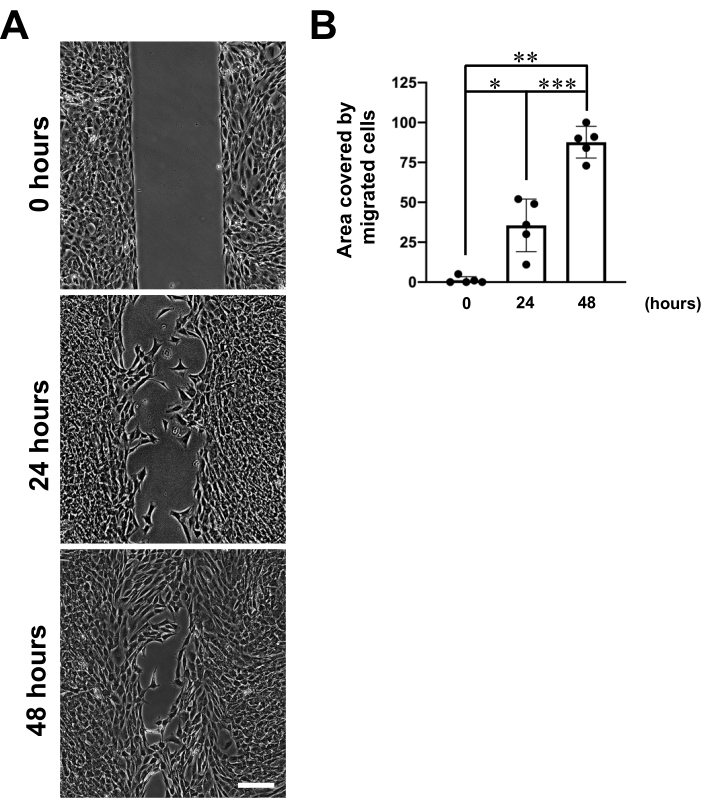
Figure 4: Representative images of O9-1 cell migration into a cell-free gap on mixed Col1/HA substrates. (A) The top panel shows the gaps at the start of the experiment (Day 0). The bottom panels show gap images after 24 h (Day 1) or 48 h (Day 2) incubation. Scale bar = 150 µm. (B) A bar graph showing the quantitative analysis of cell migration. The data represent the mean ± SD of the gap area covered by migratory cells relative to the area of the original gap (n = 5 per condition). *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired Student's t-test. Please click here to view a larger version of this figure.
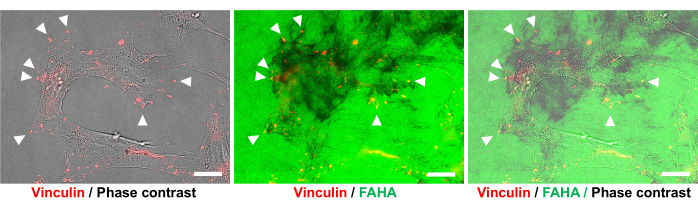
Figure 5: Degradation of HA in the mixed Col1/HA substrates at FAs in O9-1 cells. O9-1 cells cultured on mixed substrates consisting of Col1/HA were immunolabeled with anti-vinculin antibody (red). The dark spots/streaks represent HA degradation activity in the FAHA-H2 substrate (green). The sites of HA degradation and focal adhesions (vinculin) were co-localized on the mixed substrates (arrowheads). Scale bar = 500 µm. Please click here to view a larger version of this figure.
