Rat duodenal and jejunal organoids were generated using the protocol outlined in section 2. It is very important during the crypt isolation steps that villi are efficiently depleted from the PBS. If too many villi are plated in the EME with crypts, it can cause death of the entire culture and failure to establish an organoid line. Because of this, it is useful to isolate crypts under a dissecting scope, allowing for visual confirmation of villar depletion. Figure 1 depicts representative villar fragments and crypts (Figure 1A). Note the significantly smaller size of crypts compared to villi (Figure 1B). After plating, the crypts will expand into spheroids over the next few days and will begin to bud and differentiate by day 4-7 (Figure 2). Once the organoids reach an extensively budded stage, they should be passaged. During passaging, it is important to disrupt the organoids enough to split the crypt buds apart, so that organoid numbers can be expanded (Figure 3B).
The successful recovery of organoids after freezing is highly dependent on the state at which they are frozen. Organoids in a highly proliferative undifferentiated state recover with the highest efficiency. Therefore, we recommend inducing them to be spherical and cystic instead of budded and differentiated. To achieve this, Wnt can be hyperactivated by increasing the amount of the Wnt ligand R-spondin in the media and by including nicotinamide in the media, which has been shown to support organoid formation and cell survival in several culture systems30,31. Figure 3A demonstrates a healthy organoid culture just 2 days after thawing. Including BSA in the media during thawing has also helped in the survival of rat intestinal organoid cultures, which have proven to be more delicate than mouse intestinal organoids.
While 3D organoid culture is often preferred because it recapitulates some of the normal intestinal architecture, it makes other approaches, including live imaging, transfections, and lentiviral transductions, more technically challenging. The use of 2D monolayers generated from 3D organoids32 (Figure 4) allows for higher efficiency introduction of the plasmids. While 3D intestinal organoids are traditionally resistant to transient transfections, plasmids encoding for EGFP can be successfully introduced using lipid-based transfection methods. The most cost-effective approach using PEI is outlined in step 6.1 (Figure 5), but electroporation and commercially available transfection reagents have also yielded comparable results (data not shown). Future studies will be focused on whether these approaches can be used for introducing CRISPR constructs into monolayers.
It was important to be able to reform 3D organoids from 2D monolayers after transfection so that they could be maintained as a passageable line with 3D architectural components of crypts. Interestingly, 2D monolayers plated on the EME readily reformed into small spheroids when EME was added back to the top of the cells, whereas a collagen I substrate was not sufficient for the reformation of 3D structures (Figure 6).
While transient transfections are useful for many studies, the formation of stable lines are often more useful, requiring the introduction of lentivirus into the cells. Rat intestinal organoids were infected with lentivirus by modifying previously published protocols (Figure 7). A key step in the protocol is the disruption of organoids into small aggregates or cell clusters. If cultures are not efficiently disrupted and organoids remain intact, the lentiviral particles will not get into the cells. After infection, organoids must recover and regrow. The protocol outlined here allows for the uptake of viral particles by 10%-48% (mean: 19.4% ± 6.5%) of cells before selection.
Whole mount staining of organoids can prove difficult due to incomplete removal of EME residue or incomplete antibody penetrance. The protocol outlined here allows for the robust staining of organoids. The visualization of organoids on a confocal microscope can also prove difficult if they are too far away from the coverslip. By using VALAP, a well with some height is created such that organoids are not crushed by the coverslip, but are still allowed to settle close to the coverslip for ease of imaging. Representative staining against the apical anion channel cystic fibrosis transmembrane conductance regulator (CFTR) and phalloidin to label F-actin is shown in Figure 8.
Finally, organoids have utility in functional assays. Patient-derived organoids from cystic fibrosis patients have been used to screen CFTR function, as treatment with the cAMP agonist forskolin induces robust CFTR-mediated fluid secretion, causing organoid swelling29,33–37. One goal of this work was to identify and develop an organoid model that can be used in parallel to in vivo preclinical studies. Therefore, we aimed to determine whether rat intestinal organoids undergo forskolin-induced swelling. Indeed, within 30 min of forskolin treatment, rat organoids swelled, with a maximal effect observed by 120 min (Figure 9).
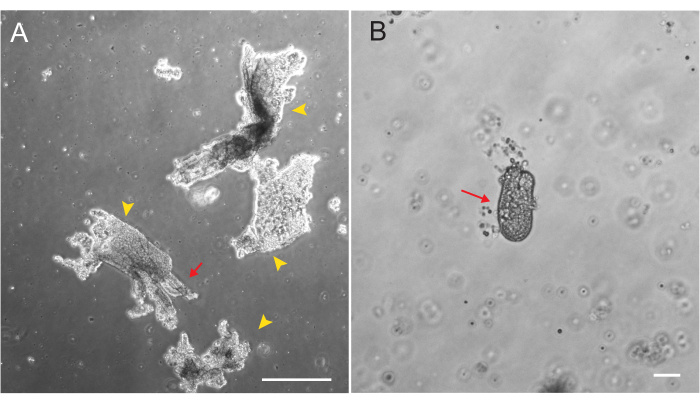
Figure 1: Villar fragments and crypts during epithelial isolation. (A) Representative image of villar fragments in EDTA solution during the crypt isolation protocol. Yellow arrowheads mark villar fragments. Red arrows depict crypts attached to a villar fragment. Note the difference in relative sizes. (B) Higher magnification image of a single crypt (red arrow) so that morphology can be visualized. Scale bars: 100 µm. Please click here to view a larger version of this figure.
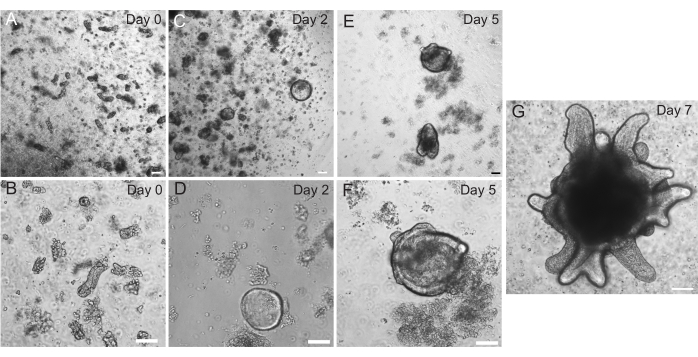
Figure 2: Rat intestinal organoid progression. Rat jejunum crypts were plated in EME immediately after isolation (A,B). Within 2 days, the crypts became spheroids (C,D). By Day 5, they began to initiate crypt buds (E,F), which elaborated and grew by Day 7 (G). Scale bars: 100 µm. Please click here to view a larger version of this figure.
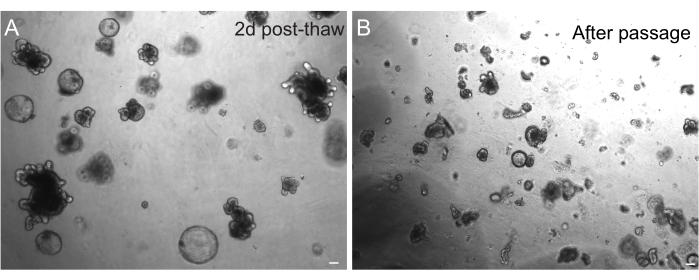
Figure 3: Organoids after thawing and passaging. (A) Rat jejunal organoids were thawed following the outlined protocols after cryopreservation. Note the presence of both spheroids and budded organoids just 2 days after thawing. (B) The same organoid line depicted in A immediately after passaging following the outlined protocol. Note the relative size difference between structures in A and B and the presence of single crypt-like domains in B. Scale bars: 100 µm. Please click here to view a larger version of this figure.
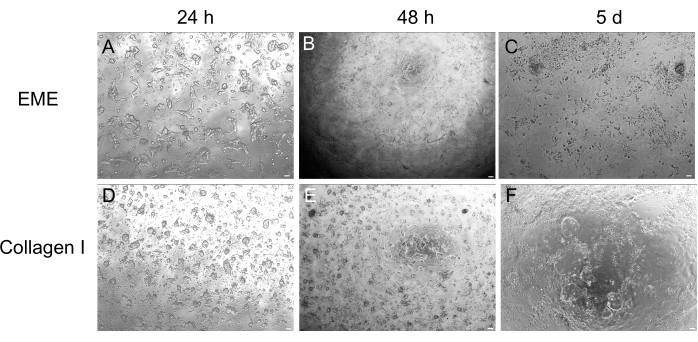
Figure 4: 2D monolayer formation from 3D organoids. (A–C) 2D monolayer progression on EME. (D–F) 2D monolayer progression on collagen I. By day 5, each condition yielded ~80% confluency. Scale bars: 100 µm. Please click here to view a larger version of this figure.
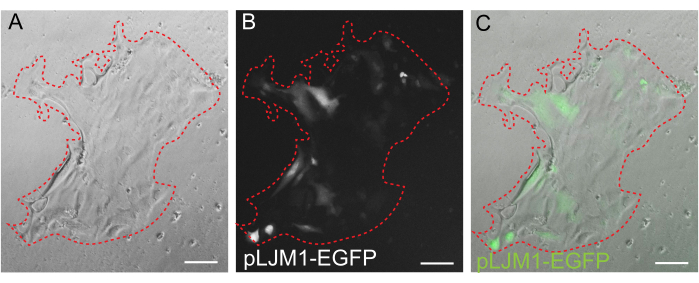
Figure 5: Transient transfection of a 2D monolayer. Representative image of a 2D monolayer grown on EME transiently transfected with pLJM1-EGFP plasmid using PEI. (A) Brightfield, (B) fluorescence (GFP), (C) overlay. The dotted red line marks the monolayer boundary. Scale bars: 100 µm. Please click here to view a larger version of this figure.
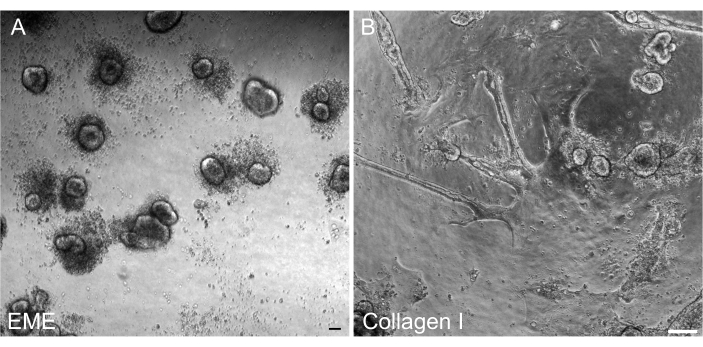
Figure 6: Reformation of 3D organoids from 2D monolayers on EME. (A) Formation of 3D organoids from 2D monolayers grown on EME. Organoids efficiently formed by 5 days after adding EME to the apical surface of the monolayer. Note the abundance of dead cells surrounding the small 3D spheroids. (B) Persistence of 2D monolayers 5 days after collagen I was added to the apical surface of 2D monolayers grown on collagen I. Scale bars: 100 µm. Please click here to view a larger version of this figure.
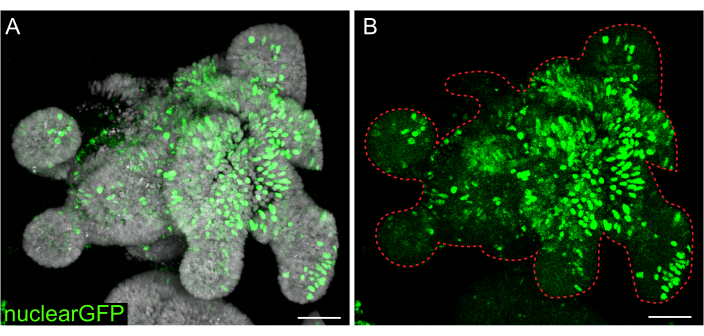
Figure 7: Lentiviral infection of 3D organoids. Rat jejunum organoids were infected with nuclear GFP lentiviral particles using the outlined protocol. After recovery and growth for 5 days, organoids were fixed and counterstained with DAPI. (A) DAPI: gray; nuclearGFP: green. (B) nuclearGFP:green. The dotted red line marks the organoid boundary. Scale bars: 50 µm. Please click here to view a larger version of this figure.
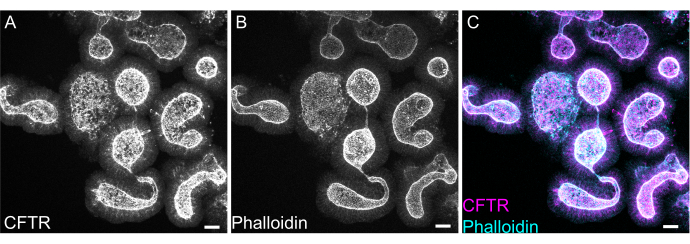
Figure 8: Whole mount immunofluorescence of rat intestinal organoids. (A) CFTR, (B) phalloidin, and (C) merged whole mount immunofluorescence of rat jejunal organoids. Note the apical enrichment of CFTR staining in organoids (gray in A, magenta in C). Phalloidin marks F- actin and prominently labels the apical brush border (gray in B, cyan in C). Scale bars: 25 µm. Please click here to view a larger version of this figure.
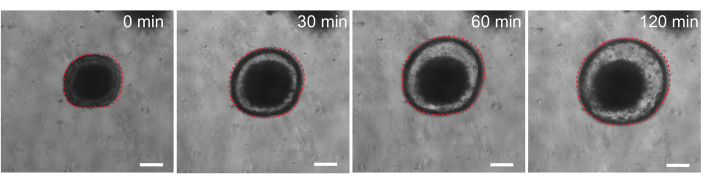
Figure 9: Rat intestinal organoids swell upon forskolin stimulation. Representative time course of rat intestinal organoid swelling after addition of the cAMP agonist forskolin. The 0 min time represents the time point immediately prior to the addition of 10 µM forskolin. Images show the same organoid at 30 min time intervals. Maximal swelling was observed at 120 min after forskolin addition. The dotted red line outlines the organoid boundary. The dark material in the middle of the organoid lumen is comprised of dead cells. Scale bars: 100 µm. Please click here to view a larger version of this figure.
Table 1: AdDMEM+ recipe. Ingredients to make the standard AdDMEM+ media, which is the base media throughout the methods shown here. Please click here to download this Table.
Table 2: Rat intestinal organoid media (rIOM) recipe. Detailed recipe of the standard rat intestinal organoid media, including solvent and storage conditions for recombinant proteins. Please click here to download this Table.
Table 3: Solutions. Recipes and instructions to make other solutions used throughout the protocol. Please click here to download this Table.
Table 4. Rat intestinal organoid medium for 2D monolayer culture (rIOM2D). Modified recipe of organoid culture media optimized for the 2D growth of monolayers. Please click here to download this Table.
