Mice were provided with water and food ad libitum and were maintained on a 12 h light/dark cycle. Mice were euthanized by exposure to isoflurane followed by cervical dislocation. All animal procedures were in accordance with the National Institutes of Health guidelines and approved by the Oregon Health and Science University Institutional Animal Care and Use Committee.
NOTE: Eye enucleation, retina dissection, and retina splitting should be performed as quickly as possible to preserve the health of the living tissue. Aim to complete the dissection in < 4 min per eye. These three steps are to be performed sequentially. Wild-type mice: Adult (>3 months) male and female C57BL/6J mice were used for experiments. For synapse morphology, mice expressing green fluorescent protein (GFP) under the Pcp2 promoter (Pcp2-cre/GFP)14 were used. Transgenic mice: For horizontal cell visualization with GFP during immunohistochemistry or electrophysiology experiments, a triple-transgenic mouse was used: vGATFLPo; vGlut2Cre; Ai80d. The vGATFlpo and vGluT2Cre strains are knock-in mice expressing Flpo or Cre recombinase downstream of their respective promoters. The Ai80d mouse is an intersectional reporter mouse (CatCh/EYFP) and will only express Ca2+ permeable channel rhodopsin (ChR2) in cells expressing Cre and Flpo recombinases. Thus, the triple transgenic mouse only expresses ChR2 in cells with a history of both VGAT and vGluT2 expression.
1. Materials preparation for retina dissection and retina splitting
- Prepare pieces of nitrocellulose membrane
NOTE: Detaching the split retina from the nitrocellulose membrane reduces background fluorescence in microscopy and simplifies patch clamp recording. Membrane removal can be performed before or after tissue fixation. For fixed split retinas, it is not necessary to treat the pieces of nitrocellulose membrane. For live split retinas, treat the membrane according to steps 1.1.3 – 1.1.5 to facilitate gentle detachment from the tissue.- Cut 16 pieces (or more) of nitrocellulose membrane into 5 mm x 5 mm squares. Extra can be prepared in bulk and stored for future use.
- Set aside half of the pieces of membrane for later use. These pieces will not be treated with a blocking solution.
- Incubate the remaining pieces in a detergent-free IHC blocking solution (such as 3% horse serum + 0.025% NaN3 diluted in PBS) for 10 min at room temperature, shaking gently.
CAUTION: Use appropriate PPE when handling NaN3, as it is a potent toxin. - Wash the membrane pieces thoroughly by incubation in bicarbonate-buffered Ames media for 10 min at room temperature, shaking gently.
- Completely air dry the blocked pieces of membrane (~20 min). Label and store the membrane pieces at room temperature, keeping them separate from the untreated pieces of membrane.
- Prepare Ames media
- Prepare bicarbonate-buffered Ames media and maintain the solution at room temperature under constant carbogenation (95% O2 and 5% CO2).
2. Mouse eye enucleation
- Euthanize the mouse by any available method according to institutional IACUC guidelines.
- Flip the mouse onto one side and use two fingers to gently press down around the eye socket. This will cause the eye to bulge out from the skull.
- Using curved dissection scissors, snip underneath the bulged eye to sever the optic nerve and to separate the eye from the skull.
- Scoop the eye with scissors and place it into a Petri dish filled with ice-cold Ames media.
NOTE: For downstream applications in which the tissue will be fixed after splitting, ice-cold PBS can be used instead of Ames media. - Repeat steps 2.1 – 2.4 for the remaining eye.
3. Retina dissection
- Use the custom glass transfer pipette to transfer one eye to a new Petri dish containing fresh, ice-cold Ames media.
NOTE: The wide opening of the custom transfer pipette prevents accidental squishing of the tissue, and using glass minimizes adhesion of the tissue to the walls of the pipette. However, a wide-mouth plastic transfer pipette is also acceptable if the experimenter is already proficient at using this tool. - Use forceps to stabilize the eye by pinning its extra connective tissue to the bottom of the Petri dish. Then, puncture the eye along the ora serrata line using a 25G needle to create an entry point for the Vannas scissors.
- Use Vannas scissors to cut along the ora serrata line until the cornea comes free from the rest of the eye (Supplementary Figure 1A). Remove the lens from the eyecup using forceps (Supplementary Figure 1B).
- Use the custom glass pipette to transfer the eyecup to a large volume (≥100 mL) of carbogenated Ames and repeat steps 3.1 – 3.3 with the remaining eye.
NOTE: Eyecups are placed into carbogenated Ames to maintain tissue health while the dissection is performed on the other eye. - Transfer one eyecup to a Petri dish filled with freshly carbogenated Ames.
- Using Vannas scissors, make a small snip inward from the edge of the sclera, then use two pairs of forceps to peel the sclera away from the retina (Supplementary Figure 1C). Avoid grabbing the retina with the forceps. Instead, pull apart the flaps of the sclera created by the scissor snip.
- Use the Vannas scissors to snip the optic nerve connecting the sclera and the retina (Supplementary Figure 1D), then gently pry the retina from the sclera using the scissors or forceps to isolate the retina. (Figure 1A).
NOTE: While the RPE will typically remain attached to the eyecup, no extra steps are required to remove the RPE in the event it is attached to the retina. At this point, the edges of the retina may be optionally trimmed with a scalpel to prevent curling during the flattening step (Figure 1B). - Use a scalpel to cut the retina into halves or quarters (Figure 1C), then use the custom transfer pipette to return the pieces to a large volume (≥ 100 mL) of continuously carbogenated Ames media.
NOTE: The choice of halves or quarters is subjective. Choose the best option for the desired application. - Repeat steps 3.5 – 3.8 for the remaining eye before proceeding to retina splitting.
4. Retina splitting
- Discard the Ames media from the Petri dishes and replace it with freshly carbogenated Ames.
NOTE: To maintain carbogenation throughout the remainder of the retina splitting procedure, replace the media in the Petri dish with freshly carbogenated Ames roughly every 5 min. - Using the custom transfer pipette, place a piece of retina onto a glass slide (7.5 cm x 5 cm), ganglion cell side up, then flatten it by removing the surrounding liquid with a delicate task wipe (Figure 1D). If necessary, gently pull the retinal edges with a fine tip paintbrush under a dissection microscope.
- Use forceps to lower a dry 5 mm x 5 mm piece of nitrocellulose membrane onto the retina, causing it to adhere to the ganglion cell side (Figure 1E).
NOTE: If membrane removal from live tissue is required (i.e., for electrophysiology), use a dry piece of serum-treated membrane for this step (see steps 1.1.3 – 1.1.5 for details). This reduces the strength of the adhesion to the ganglion cell layer, making it easier to remove the retina from the nitrocellulose post-split. - Flip the retina over so that the nitrocellulose is resting on the glass slide and place a dry piece of 5 mm x 5 mm membrane onto the photoreceptor side of the retina (Figure 1F).
- Touch the wetted tip of the paintbrush to the space between the two membranes and allow capillary action to suck the Ames into the sandwich (Figure 1G). This reduces the adherence of the membranes to the retina and is only necessary if the retina has been overly dried with the delicate task wipe.
NOTE: If the retina has lost its shiny appearance, it has been overly dried, and step 4.5 is necessary. - To ensure uniform adherence, apply light downward pressure to the upper membrane with a wet paintbrush (Figure 1H).
- While pinning the lower membrane to the glass with one pair of forceps, use a slow, steady motion to gently peel the upper membrane away with a second pair of forceps. This will cause the retina to split just above the OPL (Figure 1I).
- Discard the upper membrane containing the photoreceptors (Figure 1J, left). The lower membrane contains the inner retina, henceforth referred to as a split retina (Figure 1J, right).
- Immediately return the split retina to carbogenated Ames media.
NOTE: For experiments on living tissue, retinas may benefit from a 15-30 min recovery period in carbogenated Ames after splitting.

Figure 1: Split retina procedure. (A) Following enucleation and eyecup preparation in cold PBS or Ames media, isolate the mouse retina from the eyecup and replace the PBS with room temperature, carbogenated Ames media. (B) Using a scalpel, trim the edges of the retina away until there are no regions with an inward curl (optional). (C) Cut the retina into quarters or halves using a scalpel. (D) Place one piece of retina on a glass slide (ganglion cell side up) using the custom transfer pipette and remove all excess Ames using a delicate task wipe. Ensure that the semi-dry retina is lying flat on the glass before proceeding to the next step. Use an Ames-wetted paintbrush tip to gently unfold regions of the retina that are not flat. (E) Using forceps, place a pre-cut piece of dry nitrocellulose membrane (5 mm x 5 mm) onto the flattened retina. (F) Flip the piece of nitrocellulose so that the photoreceptor side of the retina is now facing up. Then place another dry piece of membrane onto the retina. (G) Touch the wet tip of the brush to the space between the two membranes and allow capillary action to suck the Ames into the sandwich. This reduces the adherence of the membranes to the retina and is only necessary if the retina was overly dried with the delicate task wipe. (H) Use a wet paintbrush tip to gently press downward onto the center of the sandwiched retina. (I) Use one pair of forceps to pin the bottom piece of membrane onto the glass slide, while using another pair of forceps to gently peel the top piece of membrane away from the bottom one. (J) The inner retina (left) remains on the bottom membrane while the photoreceptors (right) are pulled away with the top membrane. Panels (A), (B), (C), (D), and (J) were acquired using a dissection microscope; the scale bar represents approximately 1 mm; panels (E-I) were acquired with a smartphone camera without magnification. Please click here to view a larger version of this figure.
5. Preparation of split retinas for immunofluorescence experiments
NOTE: The split retina will still be attached to the nitrocellulose membrane until step 5.5. Complete either step 5.1, 5.2, 5.3, or 5.4, not all four as these are for different experiments.
CAUTION: Use appropriate PPE and proceed carefully when handling paraformaldehyde (fixative).
- Preparation for flatmount immunofluorescence
- Incubate the split retina in 4% paraformaldehyde on ice for 30 min using enough solution to completely cover the retina.
- Wash the split retinas 3x in 5-10 mL of room temperature PBS. Optional pause: Split retinas may be left in PBS at 4 °C for up to 24 h.
- Preparation for immunofluorescence with vertical sections of split retina
- Incubate the split retina in 4% paraformaldehyde on ice for 30 min using enough solution to completely cover the retina.
- Wash the split retinas 3x in 5-10 mL of room temperature PBS. Optional pause: Split retinas may be left in PBS at 4 °C for up to 24 h.
- With the membrane still attached, sequentially immerse the split retina in 10%, 20%, and 30% sucrose at 4 °C for 1 h each to cryoprotect the tissue.
- Embed the cryoprotected split retinas in optimal cutting temperature (O.C.T.) compound and store them at -80 °C (up to 6 months) until cryosectioning.
- Remove the embedded split retinas from -80 °C and use a cryostat to cut sections 20 µm thick. Mount the sections onto electrostatically charged glass microscope slides, allow them to air dry, then store them at -20 °C for up to 6 months.
- Preparation for dual fluorescence in situ hybridization and immunohistochemistry
- Incubate the split retina in 4% paraformaldehyde on ice for 2 h using enough solution to completely cover the retina.
- Wash the split retinas 3x in 5-10 mL of room temperature PBS. Optional pause: Split retinas may be left in PBS at 4 °C for up to 24 h.
- Preparation for electrophysiology
- Prepare patch pipettes by pulling thick-walled borosilicate glass pipettes with filament using a micropipette puller. Only use pipettes with a measured resistance between 6-10 MΩ.
- Back fill the pulled pipettes with internal solution containing (in mM): 125 K-gluconate, 8 KCl, 5 HEPES, 1 MgCl2, 1 CaCl2, 0.2 EGTA, 3 ATP-Mg, and 0.5 GTP-Na.
- Removal of the split retina from the nitrocellulose membrane
- Using a hydrophobic barrier pen, prepare circular wells on a microscope slide (~1 cm in diameter) and allow them to air dry for 5-10 min.
- Place the split retinas within the prepared hydrophobic barrier pen wells and add enough PBS to completely cover them.
- Under a dissection microscope, push the bristles of a fine paintbrush under the edges of the tissue and gently lift upward. In this manner, work around the retina in a circle to lift it away from the membrane.
- Use forceps to remove the membrane from underneath the floating piece of retina.
- Carefully aspirate away the remaining PBS so that the piece of retina comes to rest on the microscope slide, ganglion cell-side down.
NOTE: The following steps are not to be performed sequentially. Choose the appropriate protocol for the desired application (i.e., immunostaining or dual fluorescence in situ hybridization [FISH] and immunohistochemistry [IHC] or electrophysiology).
6. Immunostaining
- If not yet prepared, use a hydrophobic barrier pen to create circular wells on a microscope slide (~1 cm in diameter) and allow them to air dry for 5-10 min. All incubation steps and wash steps will be performed within these pen wells.
- Incubate the split retinas or vertical split retina sections in antibody incubation solution (AIS: 3% horse serum, 0.5% Triton X-100, 0.025% NaN3 in PBS) for 30 min at room temperature.
- Incubate the split retinas or vertical split retina sections with primary antibodies diluted in AIS for 1 h at room temperature.
NOTE: Primary antibody incubation time will require optimization for different protein targets and antibodies. - Wash the tissue 3x in room temperature PBS.
- Incubate the tissue with secondary antibodies diluted in AIS for 1 h at room temperature. Wash the tissue 3x in room temperature PBS.
- If nuclear staining is desired, incubate the tissue with DAPI diluted in PBS for 30 s at room temperature. Wash the tissue 1x in room temperature PBS.
- Apply a drop of slide mounting media to each piece of tissue and mount a glass coverslip.
- Apply nail polish around the edges of coverslip to seal the sample. Store the slide at 4 °C.
7. Dual FISH and IHC
- Bake the split retinas at 40 °C for 30 min in a hybridization oven to increase adherence to the slide.
- Complete the RNAscope FISH protocol according to the manufacturer's protocol with the following exceptions and alterations:
- No antigen retrieval step is required. Use protease III with an incubation time of 18 min at room temperature.
- Perform all wash steps on the slide within the wells made by a hydrophobic barrier pen.
- Incubate the samples in primary antibody diluted (see Table of Materials) in PBS for 30 min at 40 °C in the hybridization oven. Wash the samples 3x in room temperature PBS.
- Incubate the samples in secondary antibody diluted (see Table of Materials) in PBS for 30 min at 40 °C in the hybridization oven. Wash the samples 3x in room temperature PBS.
- Incubate the samples in 1x DAPI for 30 s at room temperature. Wash the samples 1x in room temperature PBS.
- Apply a drop of anti-fade mounting media to each piece of tissue and mount a glass coverslip.
- Apply nail polish around the edges of coverslip to seal the sample. Store the slide at 4 °C.
8. Electrophysiology
- After removing the nitrocellulose membrane, transfer a split retina into the patch-clamp recording chamber and gently anchor it in place with a platinum harp.
- Throughout the experiment, continuously perfuse the split retina with Ames solution carbogenated with 95% O2 and 5% CO2. Maintain the solution between 32-34 °C.
NOTE: During the experiment, the tissue can be visualized using Dodt gradient contrast microscopy. - Under room lighting, perform whole-cell voltage-clamping to record from INL neurons.
- While recording, simulate light responses using a microcellular injection unit to apply pharmaceutical compounds, or a 470 nm LED to stimulate channelrhodopsin (ChR2).
NOTE: Light intensity can be measured using a digital optical power meter.
- While recording, simulate light responses using a microcellular injection unit to apply pharmaceutical compounds, or a 470 nm LED to stimulate channelrhodopsin (ChR2).
9. Confocal microscopy
- For confocal immunofluorescence, take images with a confocal microscope using a 40x/1.3 or 63x/1.40 oil immersion objective. Use FIJI to adjust brightness and contrast and to generate Z-projections from image stacks.
Retina splitting preserves photoreceptor terminals
To confirm that retina splitting does not damage the dendrites of second order neurons in the OPL, vertical sections of split retinas were stained with antibodies against the synaptic vesicle protein synaptophysin (green), and protein kinase C alpha (PKCα; red). The intense band of synaptophysin labeling across the top of the split retina indicates that the photoreceptor synaptic terminals are retained (Figure 2). Furthermore, PKCα staining reveals normal morphology of rod bipolar cells (RBCs). No photoreceptor nuclei are visible, indicating that the retina is split between the OPL and innermost row of photoreceptor cell bodies (Figure 2).
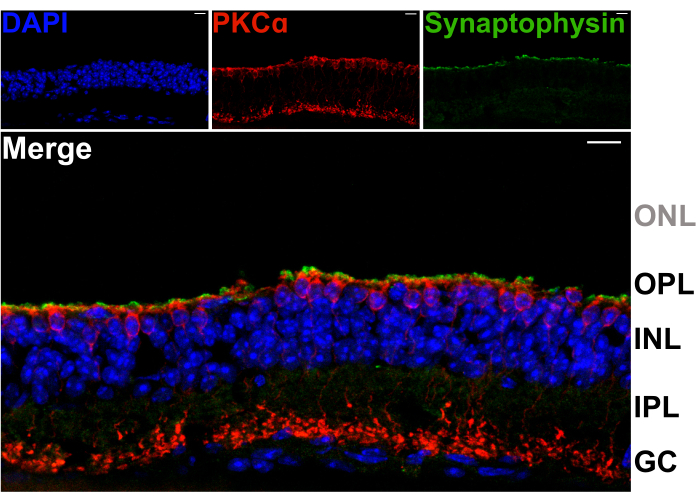
Figure 2: Split retinas retain photoreceptor terminals. Fluorescent confocal micrographs showing a vertical cross-section of a spit retina that was cryosectioned (20 µm thickness) following the splitting procedure. Each image is a maximum projection of a confocal z-stack. The section was immunolabeled with antibodies against PKCα (top center) and synaptophysin (top right) to visualize RBCs and synaptic vesicles, respectively. The merged image (bottom) shows synaptic vesicles (green), which reside in the photoreceptor terminals, just above the apical processes of the RBCs (red) in the OPL. Cell nuclei are labeled with DAPI (blue). No photoreceptor nuclei are visible within the ONL. Abbreviations: ONL = outer nuclear layer; OPL = outer plexiform layer; INL = inner nuclear layer; IPL = inner plexiform layer; GC = ganglion cells. Scale bars = 10 µm. Please click here to view a larger version of this figure.
Synapse morphology in the OPL is preserved after retina splitting
Using a mouse that expresses GFP in RBCs under the pcp2 promoter14 pre- and post-synaptic proteins in the OPL were immunolabeled to assess the integrity of this synaptic layer following a split14. Despite the shear forces occurring through the axons of the photoreceptors, splitting does not perturb the morphology of photoreceptor-BC synapses in the OPL, as normal positioning of RBC dendrites, labeled for RGS11, and photoreceptor synaptic ribbons, labeled for CtBP215 is observed (Figure 3). For each synaptic contact between rods and RBCs, RGS11 can be seen as red puncta that lie within the horseshoe shape of the synaptic ribbons (green). In a subsequent experiment, an anti-GPR179 antibody16 was used to label the post-synaptic ON-BC dendritic tips16, and an anti-PSD-95 antibody was used to label pre-synaptic rod photoreceptor terminals (Supplementary Figure 2). These results again confirm the stability of the OPL in the split retina preparation, as RBC dendrites are shown to closely associate with their pre-synaptic partner, the rod terminals.
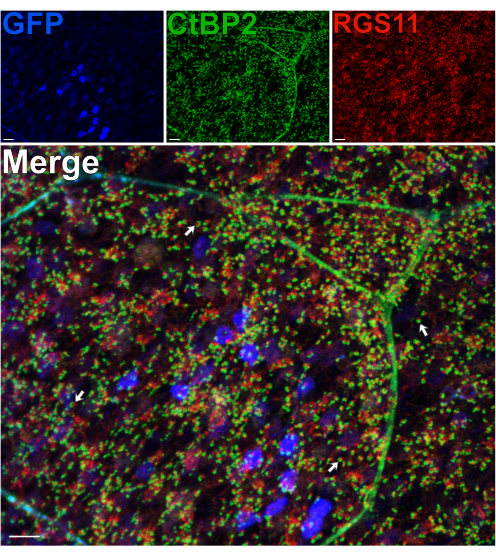
Figure 3: Synapse morphology in the OPL is preserved after retina splitting. Confocal immunofluorescence images of a split retina from a transgenic mouse expressing GFP in RBCs under the Pcp2 promoter. Levels of GFP expression (blue) vary across RBCs in the retina. Following splitting, the retina was fixed, then incubated with antibodies against CtBP2 (green) and RGS11 (red) to label photoreceptor synaptic ribbons and ON-BC dendritic tips, respectively. Each red-green pair represents a synaptic contact between a rod and an ON-BC. Scale bars = 10 µm. Please click here to view a larger version of this figure.
Retina splitting maintains RBC viability
To assess the viability of the inner-retinal neurons after a split, a membrane impermeable, near-infrared nuclear dye (MI-NIR) was used, which enables the identification of dead cells. After incubation with MI-NIR, split retinas were fixed, then labeled with anti-PKCα to identify RBCs. Confocal micrographs of the split retina reveal regional variability in cell viability across the tissue, with some regions experiencing higher rates of cell death than others. This variability may result from damage inflicted onto certain regions of the retina during the dissection, splitting, or handling procedures (Figure 4). Given that the cell bodies of RBCs reside in the outermost region of the INL, close to the site of the split, a careful evaluation of their viability was warranted. Scarce colocalization of PKCα and MI-NIR confirmed that most RBCs remain viable after retina splitting (Figure 4).
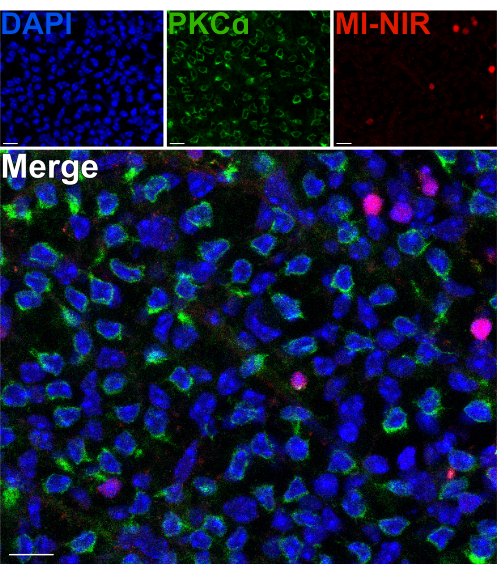
Figure 4: Rod bipolar cells are viable after retina splitting. Fluorescent confocal micrographs showing a region of a split retina in a flatmount perspective. After splitting, the live retina was incubated with MI-NIR dye (red) for 30 min at 37 °C. The retina was then fixed and immunolabeled with antibodies against PKCα to visualize RBCs. In this region of the retina, colocalization of PKCα and MI-NIR is infrequent. MI-NIR colocalizes with nuclei (blue) that do not belong to RBCs. Abbreviations: MI-NIR = membrane impermeable NIR live/dead stain. Scale bars = 10 µm. Please click here to view a larger version of this figure.
Split retinas are amenable to dual FISH and IHC
By extending the fixation time for standard IHC, split retinas can be sequentially processed by FISH and IHC to label mRNAs and proteins simultaneously17,18. Experiments confirmed that a 2 h fixation in 4% paraformaldehyde yields robust mRNA labeling while still preserving protein epitopes for antibody binding. FISH was performed on split retinas followed by IHC to visualize the expression of the GABAA receptor subunit δ (GABRD; anti-sense mRNA probes) in relation to the position of RBCs (anti-PKCα antibody) in the outer INL (Figure 5A). GABRD mRNA expression appears rare in RBCs (Figure 5A); however, the transcript is abundantly expressed by amacrine cells and ganglion cells as evidenced by the labeling pattern on transverse sections from an intact retina (Figure 5B). In the outer INL (Figure 5A), GABRD mRNA is more evenly distributed compared to the inner INL (Figure 5C) where it is concentrated in distinct cells. Antisense probes targeting other GABA receptor subunits produce distinct labeling patterns, demonstrating the specificity of the probes (data not shown).
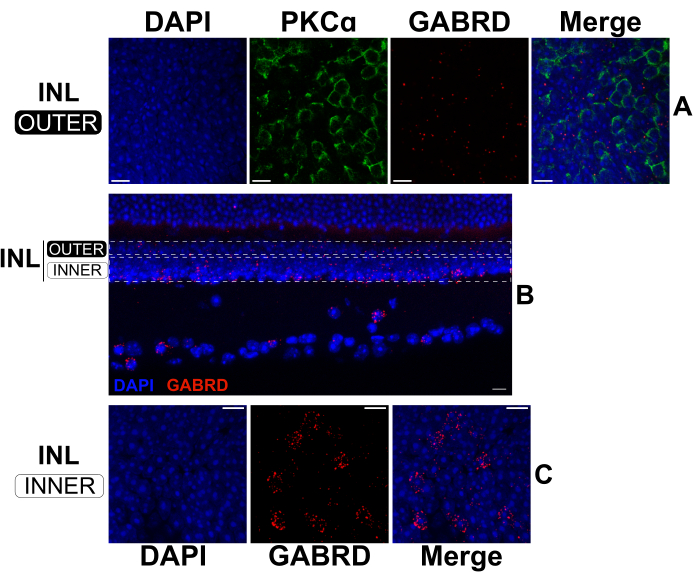
Figure 5: Dual FISH and IHC in a split retina and an intact retina. (A, C) Confocal micrographs of a flatmount split retina and (B) a vertical section from an intact retina. Images in (A) and (C) are maximum projections of optical sections in the upper and lower regions of the INL respectively. The dotted rectangles in (B) represent the approximate boundaries used to create the projections shown in (A) and (C). The split retina (A, C) was fixed for 2 h, then labeled with antisense mRNA probes against GABRD (red). Afterward, the split retina was stained with antibodies against PKCα to label RBCs (green). The PKCα channel was omitted from projections of the lower INL for clarity. The intact retina in (B) was fixed for 24 h before sectioning. Afterward, the fixed retina was labeled with antisense mRNA probes against GABRD (red). All samples were stained with DAPI (blue) for 20 s prior to coverslip mounting. Abbreviations: INL = inner nuclear layer. Scale bars= 10 µm. Please click here to view a larger version of this figure.
Split retinas are well-suited to patch-clamp electrophysiology recording from BCs and HCs
To patch a BC or HC soma in a traditional whole mount retina, the pipette must approach from either the ganglion cell side or the photoreceptor side. Both approaches require traversing several retinal layers to reach the INL, during which the pipette tip often becomes obstructed by debris. In a vibratome slice preparation, the BC and HC somas are readily accessible, but their dendritic processes may be severed, disrupting their lateral connections. In split retinas, however, the cell bodies of RBCs and HCs sit at the tissue's surface, providing greatly improved access to patch pipettes while preserving the OPL's lateral circuitry.
Figure 6 shows chemically simulated light responses recorded from BCs in a split retina. Perfused Ames medium was supplemented with L-AP4 (4 µM), a group III mGluR agonist, to simulate glutamate release from photoreceptors in darkness. The mGluR6 antagonist, CPPG (600 µM, in Ames), was puffed onto the dendrites of the patched cell (held at -60 mV) to simulate a light flash via inhibition of mGluR6. Cells responded to CPPG puffs with two types of inward currents. One type shows a transient current followed by a plateau (Figure 6A), similar to the canonical light-evoked currents recorded from RBCs in retinal slices19. The other type remains sustained throughout the puff duration (Figure 6B), resembling currents recorded from ON cone bipolar cells (ON-CBC)19.
A separate experiment was performed to target HCs, a cell type with a wide dendritic field that is often difficult to preserve in slice preparations. A mouse line expressing channel rhodopsin (ChR2) and GFP in HCs was used to facilitate easy identification under a fluorescence microscope. First, currents from HCs were recorded in response to a series of depolarization steps (-100 mV to 50 mV, step size = 15 mV) to which they responded with inward currents followed by outward currents (Figure 6C). These cells were then stimulated with a brief blue light pulse (200 ms, 470 nm) producing large, ChR2-driven inward currents in two cells (Figure 6D).
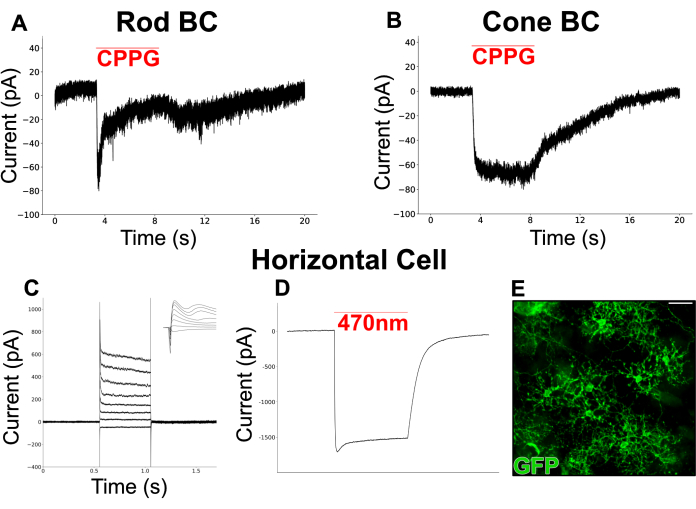
Figure 6: Patch clamp recordings from INL neurons in split retinas. (A) A putative RBC and (B) CBC were voltage-clamped at -60 mV in perfused Ames media containing L-AP4 (4 µM). Puffing CPPG (600 µM) onto the dendrites of the clamped cells invoked an inward current which was transient in the RBC but sustained in the CBC. The RBC recording in (A) is a single trace whereas the CBC recording in (B) represents the average of 3 traces. (C) A patch clamp recording from an HC in a vGATFLPo; vGlut2Cre; Ai80d mouse. The red line shows the duration of a 200 ms, 470 nm light pulse used to invoke the large, inward current through ChR2. (D) Injected current responses from an HC that was voltage clamped at -60 mV, then stepped between -70 mV and +35 mV in 15 mV intervals and returned to -60 mV. The inset shows the same traces in a 6 ms window surrounding the start of the voltage step. (E) Immunofluorescent micrograph of a flatmount split retina showing horizontal cells expressing GFP in a vGATFLPo; vGlut2Cre; Ai80d mouse. Scale bar = 20 µm. Electrophysiology data were collected at a 20 kHz sampling rate and filtered with a low-pass Bessel filter at 5 kHz. Data were then exported, and offline visualization and analysis were performed using Python 3. Please click here to view a larger version of this figure.
Retina splitting enables rapid interrogation of INL and OPL anatomy
The retina's external limiting membrane (ELM) and ONL comprise a barrier ~90 µm thick, which impedes the diffusion of antibodies into the inner retina and creates suboptimal immunostaining conditions20,21,22. Therefore, immunolabeling targets in the OPL or INL using a conventional flatmount retina requires time-intensive staining protocols that often necessitate 48-96 h antibody incubations5,6,7,8,20,22.
Removing the photoreceptors allows for rapid antibody penetration of inner retinal neurons. As a result, labeling of inner-retina protein targets can be achieved in as little as 1 h with the use of dye-conjugated primary antibodies. Antibodies against PKCα and Calbindin-D were used to label RBCs and HCs of the INL respectively (Figure 7). Unlike traditional vertical retina sections that truncate the lateral processes of wide-field neurons, the split retina preparation enables visualization of the full dendritic arbor of wide-field cells such as HCs (Figure 6E, Figure 7).
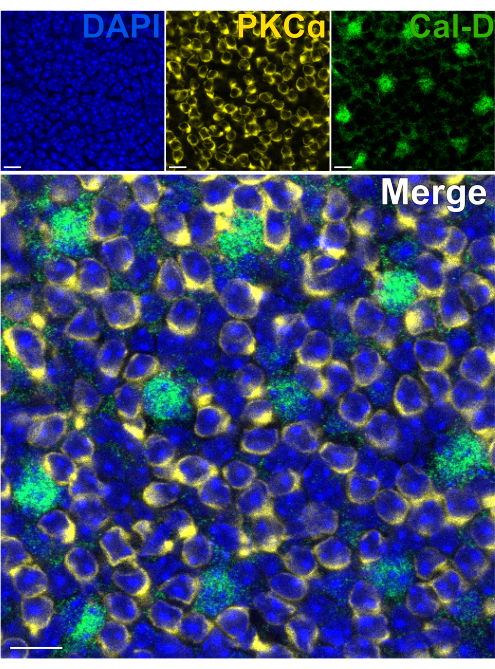
Figure 7: Rapid immunolabeling of inner retina proteins in a split retina. Confocal immunofluorescence images of a split retina from a flatmount perspective. The split retina was incubated with antibodies against PKCα (yellow) and Calbindin-D (green) for 1 h at room temperature to label ON-BCs and HCs respectively. (A) Each single channel image is an average Z-projection composed of four optical sections: DAPI, Average z10-13; Calbindin-D, Average z11-14; PKCα, Average z11-14. (B) In the merged image, the same projections are superimposed. Scale bars = 10 µm. Please click here to view a larger version of this figure.
Supplementary Figure 1: Key stages of the retina dissection. All images were taken with a smartphone camera mounted to the ocular lenses of a dissection microscope. (A) A top-down image of a mouse eye following removal of the cornea. (B) A top-down image of the mouse eyecup after the lens has been removed. (C) A small incision is made in the sclera on the mouse eyecup. Arrows indicate the two flaps of the sclera which are pulled in opposite directions by forceps to begin separating the retina from the sclera. (D) After the sclera has been partially pulled away from the retina, Vannas scissors are inserted between the sclera and the retina, and the optic nerve is severed, freeing the retina. The red dotted circle shows the optic nerve head, and the scissors demonstrate the correct cutting trajectory (insert scissors between the sclera and the retina). The isolated retina after the sclera is pried away. Please click here to download this File.
Supplementary Figure 2: Characterization of pre- and post-synaptic components of the OPL in the split retina. Confocal immunofluorescence images from the OPL in a split retina. The split retina was incubated with antibodies against GPR179 and PSD95 for 1 h at room temperature to label to the dendritic tips of ON-BCs and the terminals of rod photoreceptors, respectively. The left and center images are maximum projections of several optical sections; the same projections are superimposed in the rightmost image. GPR179 puncta in the ON-BC dendritic tips are seen to associate closely with the rod photoreceptor terminals, demonstrating intact synaptic contacts within the OPL. Scale bars = 10 µm. Please click here to download this File.
Supplementary Figure 3: Troubleshooting: assessing the quality of a split retina. Fluorescent micrographs of a split retina stained with DAPI to visualize cell nuclei. Cells can be identified based on the diameter and tissue depth of the nucleus. (A) Photoreceptor nuclei are smaller, brighter, and more superficial, whereas (B) BC nuclei are larger, dimmer, and deeper. (C) A low magnification image of a region where photoreceptors were incompletely removed. The nuclei that appear in focus are from BCs, which are deeper than the photoreceptor nuclei on the edges of the image that appear out of focus. Scale bars for (A) and (B) = 20 µm. Scale bar for (C) = 50 µm. Please click here to download this File.
