Figure 2 provides a simplified illustration showing the sequence of fluorescence changes expected following the successful stimulation of sperm. The top panel of Figure 2 illustrates the changes in GCaMP3 fluorescence intensity, where the signal is initially dim (baseline acrosomal calcium concentrations are lower than GCaMP3 KD), and upon the entry of calcium ions via fusion pores, the fluorescence increases in brightness. Finally, upon AE, there is a loss of signal due to diffusion and the loss of the sensor to the extracellular space. The bottom panel of Figure 2 illustrates the changes in mCherry fluorescence intensity, where the initial signal is bright, and only upon AE and diffusion of the sensor along with other contents of the acrosomal matrix, there is a dimming of the red signal.
Figure 3 provides actual measured data for the stages illustrated above in two live sperm stimulated with 125 µM of the ganglioside GM1 (for more details on how GM1 regulates AE in sperm12), captured in a single field of view. Figure 3A,B, and Figure 3E,F show the red and green signals measured in two sperm at time point 0 (before stimulation), where initially, the mCherry signal is bright, and the GCaMP3 is dim. Figure 3C, Figure 3G, and Figure 3D, Figure 3H provide the Z Profiler analysis for the full duration of the experiment (raw data or F/F0, respectively). Insert panels provide a zoomed-in view of the time window where ACR and AE occur.
Because sperm are both highly heterogeneous and very sensitive to membrane damage due to various handling procedures, Figure 5 provides examples of two types of negative results. Figure 5A,C show several sperm captured within one field of view. The yellow outline highlights two sperm cells that demonstrate a high GCaMP3 signal at time point 0 (before stimulation). In the present experiments, such cells were avoided, as they indicate that the acrosomal vesicle is already going through some membrane fusion process or that they have experienced some membrane damage that allows calcium to leak. The sperm in Figure 5A indicated by the arrow (circle ROI in Figure 5A,C) demonstrates an initial mCherry signal but no response in the red or green channels (Figure 5B,D, respectively, showing the Z-Profiler analysis window as provided by ImageJ) following stimulation with 125 µM GM1. Although a subpopulation of sperm, such as the one provided in this example, shows no response to stimulation, these are included in the final analysis and are calculated in the percent response data analysis2 (Figure 3).
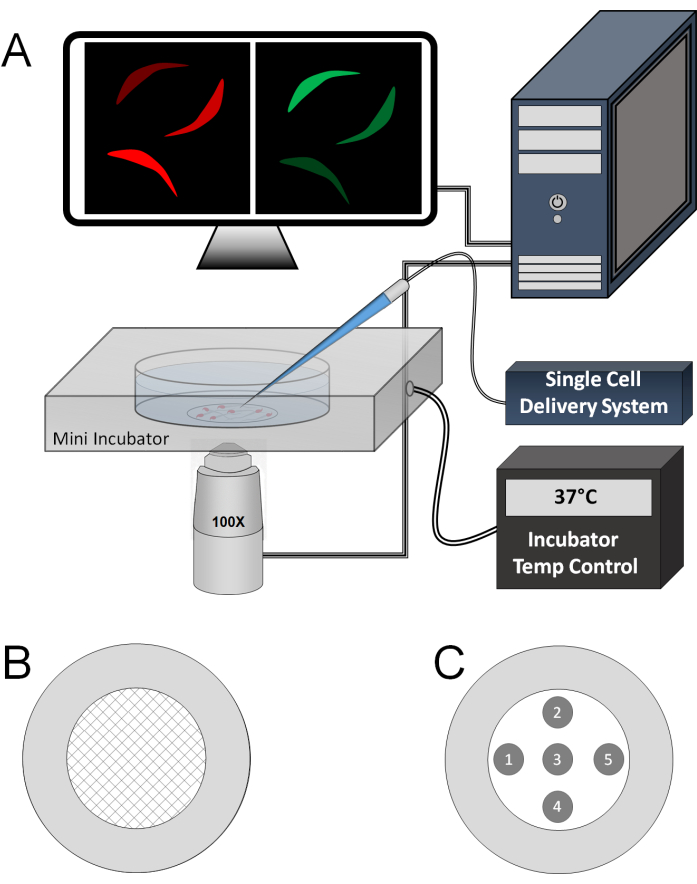
Figure 1: Instrumentation and experimental setup. (A) Live sperm cells on a PDL-coated coverslip dish placed in an incubation chamber at 37 °C. A borosilicate capillary, pulled and attached to a single-cell delivery system, is positioned near the sperm cell in the center of the imaging area for direct stimulant delivery. Both green (GCaMP3) and red (mCherry) channels are continuously monitored and recorded at a high frame rate (e.g., 10 frames/s) during stimulation. (B) Schematic representation of the PDL deposition pattern on the cover-slide dish. (C) Schematic representation of the specific areas on the dish where sperm can be stimulated using the capillary to minimize non-specific stimulation from prior puffs in the same dish. Please click here to view a larger version of this figure.
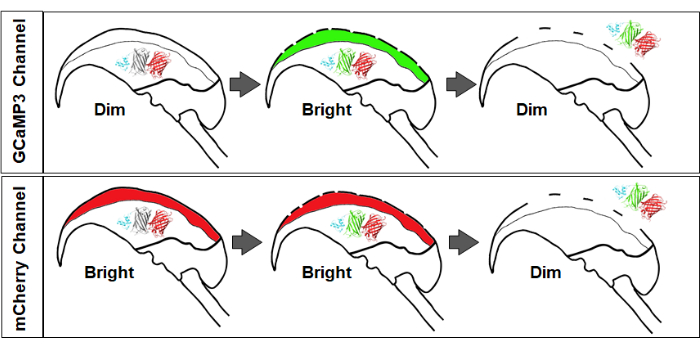
Figure 2: Changes in fluorescence signal and intensity. Top panel: An increase in GCaMP3 signal signifies calcium influx into the acrosome, followed by signal loss due to diffusion of the AcroSensE fusion protein. Bottom panel: mCherry signal remains stable during initial membrane fusion, with subsequent dimming upon AcroSensE diffusion and acrosomal exocytosis (AE). Please click here to view a larger version of this figure.
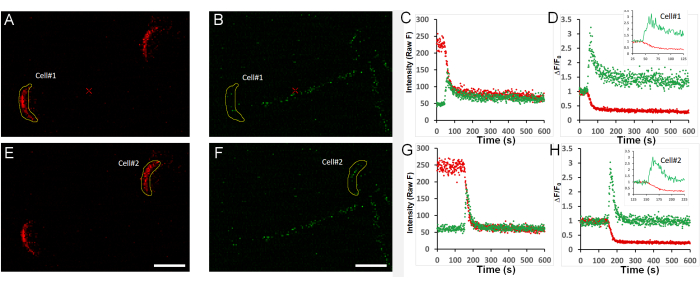
Figure 3: Representative data and fluorescence traces. A field containing two sperm cells was imaged. Green/red channel fluorescence was quantified offline using ImageJ and plotted in Excel (as described in step 6). (A–D) Quantification of red (A) and green (B) channel fluorescence intensity for cell #1 over time, calculated for selected ROIs, and then presented as raw data (C) or normalized to the initial signal (D, F/F0). The insert in (D) provides a zoomed-in view of the time frame from 25-125 s. (E–H) Quantification of red (E) and green (F) channel fluorescence intensity for cell #2 over time, calculated for selected ROIs, and then presented as raw data (G) or normalized to the initial signal (H, F/F0). The insert in (H) provides a zoomed-in view of the time frame from 25-125 s. Scale bar = 5 µm. All x-axes represent time in seconds (s). Please click here to view a larger version of this figure.
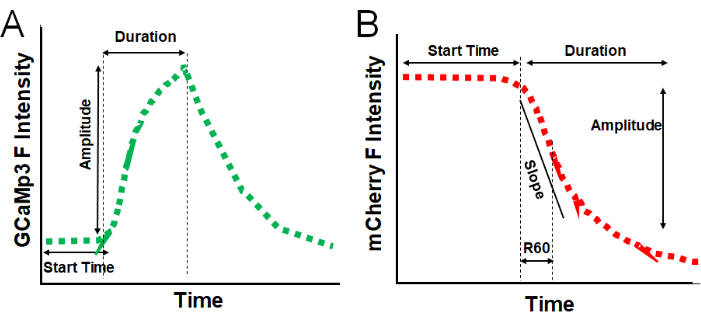
Figure 4: Data analysis and quantification parameters. (A) Illustration of a typical GCaMP3 fluorescence intensity trace, displaying quantifiable parameters, including Start Time (seconds), Amplitude (fluorescence intensity), and Duration (seconds). (B) Illustration of a typical mCherry fluorescence intensity trace, displaying quantifiable parameters, including Start Time (seconds), Duration (seconds), Slope (F/s), R60 (fluorescence intensity), and Amplitude (fluorescence intensity). Please click here to view a larger version of this figure.
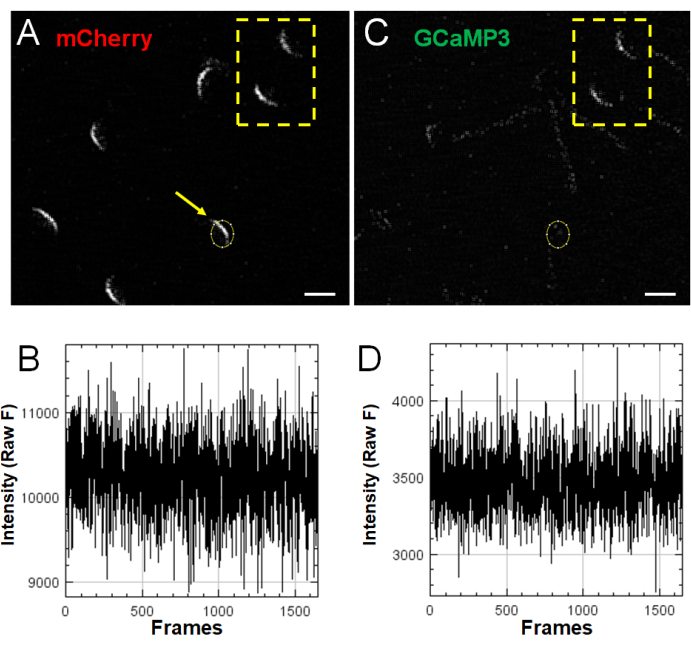
Figure 5: Negative results. (A,C) mCherry (red) and GCaMP3 (green) channels showing multiple sperm cells captured in a single field of view. The arrow in (A) indicates a sperm cell initially exhibiting mCherry fluorescence but no subsequent fluorescence intensity changes in response to 125 µM GM1 stimulation, as observed in the Z-Profiler generated traces provided in (B) (mCherry) and (D) (GCaMP3). Cells within the yellow rectangle exhibit elevated GCaMP3 fluorescence intensity at time point 0, before stimulation. Scale bar = 5 µm. Please click here to view a larger version of this figure.
