Transkriptom består af ekspressionerne af alle generne i en prøve og kan profileres af teknologier med høj kapacitet som microarray og RNA-seq1. Ekspressionsniveauerne for et gen i et datasæt kaldes en transkriptomisk funktion, og differentiel repræsentation af en transkriptomisk funktion mellem fænotypen og kontrolgrupperne definerer dette gen som en biomarkør for denne fænotype 2,3. Transkriptomiske biomarkører er blevet anvendt i vid udstrækning i undersøgelserne af sygdomsdiagnose4, biologisk mekanisme5 og overlevelsesanalyse 6,7 osv.
Genaktivitetsmønstre i det sunde væv bærer afgørende information om livet 8,9. Disse mønstre giver uvurderlig indsigt og fungerer som ideelle referencer til forståelse af de komplekse udviklingsbaner for godartede lidelser10,11 og dødelige sygdomme12. Gener interagerer med hinanden, og transkriptomer repræsenterer de endelige ekspressionsniveauer efter deres komplicerede interaktioner. Sådanne mønstre formuleres som transkriptionsreguleringsnetværk13 og metabolismenetværk14 osv. Ekspressionerne af messenger-RNA’er (mRNA’er) kan transkriptionelt reguleres af transkriptionsfaktorer (TF’er) og lange intergene ikke-kodende RNA’er (lincRNA’er)15,16,17. Konventionel differentialekspressionsanalyse ignorerede sådanne komplekse geninteraktioner med antagelsen om uafhængighed mellem funktioner18,19.
Nylige fremskridt inden for grafneurale netværk (GNN’er) viser ekstraordinært potentiale i at udtrække vigtig information fra OMIC-baserede data til kræftstudier20, f.eks. identifikation af co-ekspressionsmoduler21. GNN’ernes medfødte kapacitet gør dem ideelle til modellering af de indviklede forhold og afhængigheder mellem gener22,23.
Biomedicinske undersøgelser fokuserer ofte på nøjagtigt at forudsige en fænotype mod kontrolgruppen. Sådanne opgaver er almindeligvis formuleret som binære klassifikationer 24,25,26. Her er de to klasseetiketter typisk kodet som 1 og 0, sandt og falsk, eller endda positivt og negativt27.
Denne undersøgelse havde til formål at tilvejebringe en brugervenlig protokol til generering af transkriptionsreguleringsvisningen (mqTrans) af et transkriptomdatasæt baseret på den forududdannede grafopmærksomhedsnetværk (GAT) referencemodel. Multitask-GAT-rammen fra et tidligere offentliggjort værk26 blev brugt til at transformere transkriptomiske funktioner til mqTrans-funktionerne. Et stort datasæt af sunde transkriptomer fra University of California, Santa Cruz (UCSC) Xena platform28 blev brugt til at prætræne referencemodellen (HealthModel), som kvantitativt målte transkriptionsreglerne fra regulatoriske faktorer (TF’er og lincRNA’er) til mål-mRNA’erne. Den genererede mqTrans-visning kan bruges til at opbygge forudsigelsesmodeller og registrere mørke biomarkører. Denne protokol bruger patientdatasættet for kolonadenocarcinom (COAD) fra The Cancer Genome Atlas (TCGA) database29 som et illustrativt eksempel. I denne sammenhæng kategoriseres patienter i trin I eller II som negative prøver, mens de i trin III eller IV betragtes som positive prøver. Fordelingen af mørke og traditionelle biomarkører på tværs af de 26 TCGA-kræfttyper sammenlignes også.
Beskrivelse af HealthModel-pipelinen
Den metode, der anvendes i denne protokol, er baseret på den tidligere offentliggjorte ramme26 som skitseret i figur 1. Til at begynde med skal brugerne forberede inputdatasættet, indføre det i den foreslåede HealthModel-pipeline og hente mqTrans-funktioner. Detaljerede instruktioner til dataforberedelse findes i afsnit 2 i protokolafsnittet. Derefter har brugerne mulighed for at kombinere mqTrans-funktioner med de originale transkriptomiske funktioner eller kun fortsætte med de genererede mqTrans-funktioner. Det producerede datasæt underkastes derefter en proces til valg af funktioner, hvor brugerne har fleksibiliteten til at vælge deres foretrukne værdi for k i k-fold krydsvalidering til klassificering. Den primære evalueringsmetrik, der anvendes i denne protokol, er nøjagtighed.
HealthModel26 kategoriserer de transkriptomiske egenskaber i tre forskellige grupper: TF (transkriptionsfaktor), lincRNA (langt intergent ikke-kodende RNA) og mRNA (messenger-RNA). TF-funktionerne er defineret ud fra de annoteringer, der er tilgængelige i Human Protein Atlas 30,31. Dette arbejde udnytter annotationerne af lincRNA’er fra GTEx-datasættet32. Gener, der tilhører veje på tredje niveau i KEGG-databasen33, betragtes som mRNA-funktioner. Det er værd at bemærke, at hvis en mRNA-funktion udviser regulatoriske roller for et målgen som dokumenteret i TRRUST-databasen34, omklassificeres det til TF-klassen.
Denne protokol genererer også manuelt de to eksempelfiler for gen-id’erne for regulatoriske faktorer (regulatory_geneIDs.csv) og mål-mRNA (target_geneIDs.csv). Den parvise afstandsmatrix blandt de regulatoriske egenskaber (TF’er og lincRNA’er) beregnes ved hjælp af Pearson-korrelationskoefficienterne og grupperes af den populære værktøjsvægtede gen-co-ekspressionsnetværksanalyse (WGCNA)36 (adjacent_matrix.csv). Brugere kan direkte bruge HealthModel-pipelinen sammen med disse eksempelkonfigurationsfiler til at generere mqTrans-visningen af et transkriptomisk datasæt.
Tekniske detaljer om HealthModel
HealthModel repræsenterer de indviklede forhold mellem TF’er og lincRNA’er som en graf, hvor inputfunktionerne tjener som hjørnerne betegnet med V og en inter-vertex kantmatrix udpeget som E. Hver prøve er kendetegnet ved K-regulatoriske træk, symboliseret som VK×1. Specifikt omfattede datasættet 425 TF’er og 375 lincRNA’er, hvilket resulterede i en prøvedimensionalitet på K = 425 + 375 = 800. For at etablere kantmatrixen E anvendte dette arbejde det populære værktøj WGCNA35. Den parvise vægt, der forbinder to hjørner repræsenteret som 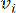 og
og 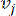 , bestemmes af Pearson-korrelationskoefficienten. Det genregulerende netværk udviser en skalafri topologi36, der er kendetegnet ved tilstedeværelsen af hubgener med centrale funktionelle roller. Vi beregner korrelationen mellem to funktioner eller hjørner og ved hjælp af det topologiske overlapningsmål (TOM) som følger:
, bestemmes af Pearson-korrelationskoefficienten. Det genregulerende netværk udviser en skalafri topologi36, der er kendetegnet ved tilstedeværelsen af hubgener med centrale funktionelle roller. Vi beregner korrelationen mellem to funktioner eller hjørner og ved hjælp af det topologiske overlapningsmål (TOM) som følger:
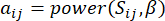 (1)
(1)
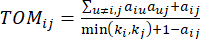 (2)
(2)
Den bløde tærskel β beregnes ved hjælp af funktionen ‘pickSoft Threshold’ fra WGCNA-pakken. Effekteksponentialfunktionen aij anvendes, hvor 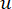 repræsenterer et gen eksklusive i og j og
repræsenterer et gen eksklusive i og j og 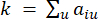 repræsenterer toppunktforbindelsen. WGCNA grupperer ekspressionsprofilerne for de transkriptomiske træk i flere moduler ved hjælp af et almindeligt anvendt ulighedsmål (
repræsenterer toppunktforbindelsen. WGCNA grupperer ekspressionsprofilerne for de transkriptomiske træk i flere moduler ved hjælp af et almindeligt anvendt ulighedsmål (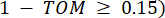 37.
37.
HealthModel-rammen blev oprindeligt designet som en multitask-læringsarkitektur26. Denne protokol bruger kun modelfortræningsopgaven til konstruktion af den transkriptomiske mqTrans-visning. Brugeren kan vælge at forfine den forudtrænede HealthModel yderligere under opmærksomhedsnetværket for multitask-grafer med yderligere opgavespecifikke transkriptomiske prøver.
Tekniske detaljer om valg og klassificering af funktioner
Funktionsvalgspuljen implementerer elleve FS-algoritmer (feature selection). Blandt dem er tre filterbaserede FS-algoritmer: valg af K bedste funktioner ved hjælp af den maksimale informationskoefficient (SK_mic), valg af K-funktioner baseret på FPR for MIC (SK_fpr) og valg af K-funktioner med den højeste falske opdagelsesrate for MIC (SK_fdr). Derudover vurderer tre træbaserede FS-algoritmer individuelle funktioner ved hjælp af et beslutningstræ med Gini-indekset (DT_gini), adaptive boostede beslutningstræer (AdaBoost) og tilfældig skov (RF_fs). Puljen indeholder også to indpakningsmetoder: Rekursiv funktionseliminering med lineær støttevektorklassifikator (RFE_SVC) og eliminering af rekursiv funktion med den logistiske regressionsklassifikator (RFE_LR). Endelig er to integreringsalgoritmer inkluderet: lineær SVC-klassifikator med de højest rangerede L1-funktionsvigtighedsværdier (lSVC_L1) og logistisk regressionsklassifikator med de højest rangerede L1-funktionsvigtighedsværdier (LR_L1).
Klassifikatorpuljen anvender syv forskellige klassifikatorer til at opbygge klassificeringsmodeller. Disse klassifikatorer omfatter lineær støttevektormaskine (SVC), Gaussisk naiv Bayes (GNB), logistisk regressionsklassifikator (LR), k-nærmeste nabo, med k indstillet til 5 som standard (KNN), XGBoost, tilfældig skov (RF) og beslutningstræ (DT).
Den tilfældige opdeling af datasættet i toget: testundersæt kan indstilles på kommandolinjen. Det demonstrerede eksempel bruger forholdet mellem tog: test = 8: 2.
