Transkriptom består av uttrycken av alla gener i ett prov och kan profileras med hjälp av tekniker med hög genomströmning som mikroarray och RNA-seq1. Uttrycksnivåerna för en gen i en datauppsättning kallas en transkriptomisk egenskap, och den differentiella representationen av en transkriptomisk egenskap mellan fenotypen och kontrollgruppen definierar denna gen som en biomarkör för denna fenotyp 2,3. Transkriptomiska biomarkörer har använts i stor utsträckning i undersökningar av sjukdomsdiagnos4, biologisk mekanism5 och överlevnadsanalys 6,7, etc.
Genaktivitetsmönster i de friska vävnaderna bär på avgörande information om livet 8,9. Dessa mönster ger ovärderliga insikter och fungerar som idealiska referenser för att förstå de komplexa utvecklingsbanorna för godartade sjukdomar10,11 och dödliga sjukdomar12. Gener interagerar med varandra, och transkriptom representerar de slutliga uttrycksnivåerna efter deras komplicerade interaktioner. Sådana mönster är formulerade som transkriptionellt regleringsnätverk13 och metabolismnätverk14, etc. Uttrycken av budbärar-RNA (mRNA) kan transkriptionellt regleras av transkriptionsfaktorer (TF) och långa intergena icke-kodande RNA (lincRNA)15,16,17. Konventionell differentiell uttrycksanalys ignorerade sådana komplexa geninteraktioner med antagandet om oberoende mellan egenskaper18,19.
De senaste framstegen inom grafneurala nätverk (GNN) visar en extraordinär potential när det gäller att extrahera viktig information från OMIC-baserade data för cancerstudier20, t.ex. genom att identifiera kouttrycksmoduler21. GNN:s medfödda kapacitet gör dem idealiska för att modellera de intrikata relationerna och beroendena mellan gener22,23.
Biomedicinska studier fokuserar ofta på att exakt förutsäga en fenotyp mot kontrollgruppen. Sådana uppgifter formuleras vanligtvis som binära klassificeringar 24,25,26. Här kodas de två klassetiketterna vanligtvis som 1 och 0, sant och falskt, eller till och med positivt och negativt27.
Denna studie syftade till att tillhandahålla ett lättanvänt protokoll för att generera transkriptionsregleringen (mqTrans) av en transkriptomdatauppsättning baserad på den förtränade referensmodellen för graf-uppmärksamhetsnätverk (GAT). Multitask-GAT-ramverket från ett tidigare publicerat arbete26 användes för att transformera transkriptomiska funktioner till mqTrans-funktionerna. En stor datamängd av friska transkriptom från University of California, Santa Cruz (UCSC) Xena-plattform28 användes för att förträna referensmodellen (HealthModel), som kvantitativt mätte transkriptionsreglerna från de regulatoriska faktorerna (TF och lincRNA) till mål-mRNA. Den genererade mqTrans-vyn kan användas för att bygga prediktionsmodeller och upptäcka mörka biomarkörer. Detta protokoll använder patientdatasetet colon adenocarcinom (COAD) från databasen The Cancer Genome Atlas (TCGA)29 som ett illustrativt exempel. I detta sammanhang kategoriseras patienter i stadium I eller II som negativa prover, medan de i stadium III eller IV betraktas som positiva prover. Fördelningen av mörka och traditionella biomarkörer mellan de 26 TCGA-cancertyperna jämförs också.
Beskrivning av HealthModel-pipelinen
Den metod som används i detta protokoll bygger på det tidigare offentliggjorda ramverket26, som beskrivs i figur 1. Till att börja med måste användarna förbereda indatauppsättningen, mata in den i den föreslagna HealthModel-pipelinen och hämta mqTrans-funktioner. Detaljerade instruktioner för förberedelse av data finns i avsnitt 2 i protokollavsnittet. Därefter har användarna möjlighet att kombinera mqTrans-funktioner med de ursprungliga transkriptomiska funktionerna eller endast fortsätta med de genererade mqTrans-funktionerna. Den producerade datauppsättningen utsätts sedan för en funktionsvalsprocess, där användarna har flexibiliteten att välja önskat värde för k i k-faldig korsvalidering för klassificering. Det primära utvärderingsmåttet som används i detta protokoll är noggrannhet.
HealthModel26 kategoriserar de transkriptomiska egenskaperna i tre distinkta grupper: TF (transkriptionsfaktor), lincRNA (långt intergent icke-kodande RNA) och mRNA (budbärar-RNA). TF-egenskaperna definieras baserat på de annoteringar som finns tillgängliga i Human Protein Atlas30,31. Detta arbete använder annoteringar av lincRNA från GTEx-datasetet32. Gener som tillhör den tredje nivåns vägar i KEGG-databasen33 betraktas som mRNA-egenskaper. Det är värt att notera att om en mRNA-funktion uppvisar reglerande roller för en målgen som dokumenterats i TRRUST-databasen34, omklassificeras den till TF-klassen.
Detta protokoll genererar också manuellt de två exempelfilerna för gen-ID:n för regulatoriska faktorer (regulatory_geneIDs.csv) och mål-mRNA (target_geneIDs.csv). Den parvisa avståndsmatrisen mellan de regulatoriska egenskaperna (TF och lincRNA) beräknas med hjälp av Pearsons korrelationskoefficienter och klustras med hjälp av den populära verktygsviktade nätverksanalysen för genuttryck (WGCNA)36 (adjacent_matrix.csv). Användare kan direkt använda HealthModel-pipelinen tillsammans med dessa exempelkonfigurationsfiler för att generera mqTrans-vyn för en transkriptomisk datauppsättning.
Teknisk information om HealthModel
HealthModel representerar de intrikata relationerna mellan TF och lincRNA som en graf, där indatafunktionerna fungerar som hörnen som betecknas med V och en gränsmatris mellan hörnen som betecknas som E. Varje prov kännetecknas av K-reglerande egenskaper, symboliserade som VK×1. Specifikt omfattade datauppsättningen 425 TF och 375 lincRNA, vilket resulterade i en provdimensionalitet på K = 425 + 375 = 800. För att fastställa kantmatrisen E användes det populära verktyget WGCNA35 i detta arbete. Den parvisa vikten som länkar två hörn representerade som 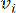 och
och 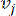 , bestäms av Pearsons korrelationskoefficient. Det genreglerande nätverket uppvisar en skalfri topologi36, kännetecknad av närvaron av navgener med centrala funktionella roller. Vi beräknar korrelationen mellan två objekt eller hörn , och , med hjälp av det topologiska överlappningsmåttet (TOM) enligt följande:
, bestäms av Pearsons korrelationskoefficient. Det genreglerande nätverket uppvisar en skalfri topologi36, kännetecknad av närvaron av navgener med centrala funktionella roller. Vi beräknar korrelationen mellan två objekt eller hörn , och , med hjälp av det topologiska överlappningsmåttet (TOM) enligt följande:
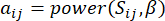 (1)
(1)
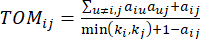 (2)
(2)
Det mjuka tröskelvärdet β beräknas med hjälp av funktionen “pickSoft Threshold” från WGCNA-paketet. Den exponentiella potensfunktionen aij tillämpas, där 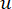 representerar en gen exklusive i och j, och
representerar en gen exklusive i och j, och 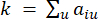 representerar vertexkonnektiviteten. WGCNA klustrar uttrycksprofilerna för de transkriptomiska funktionerna i flera moduler med hjälp av ett vanligt olikhetsmått (
representerar vertexkonnektiviteten. WGCNA klustrar uttrycksprofilerna för de transkriptomiska funktionerna i flera moduler med hjälp av ett vanligt olikhetsmått (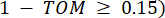 37.
37.
HealthModel-ramverket utformades ursprungligen som en arkitektur för multitasking-inlärning26. Det här protokollet använder endast modellens förträningsuppgift för konstruktionen av den transkriptomiska mqTrans-vyn. Användaren kan välja att ytterligare förfina den förtränade HealthModel under multitask-grafens uppmärksamhetsnätverk med ytterligare uppgiftsspecifika transkriptomiska exempel.
Tekniska detaljer om val och klassificering av funktioner
Funktionsvalspoolen implementerar elva FS-algoritmer (feature selection). Bland dem är tre filterbaserade FS-algoritmer: välja K bästa funktioner med hjälp av maximal informationskoefficient (SK_mic), välja K-funktioner baserat på FPR för MIC (SK_fpr) och välja K-funktioner med den högsta falska upptäcktsfrekvensen för MIC (SK_fdr). Dessutom utvärderar tre trädbaserade FS-algoritmer enskilda funktioner med hjälp av ett beslutsträd med Gini-index (DT_gini), adaptiva förstärkta beslutsträd (AdaBoost) och slumpmässig skog (RF_fs). Poolen innehåller också två omslutningsmetoder: Eliminering av rekursiva funktioner med den linjära stödvektorklassificeraren (RFE_SVC) och eliminering av rekursiva funktioner med den logistiska regressionsklassificeraren (RFE_LR). Slutligen ingår två inbäddningsalgoritmer: linjär SVC-klassificerare med de högst rankade L1-funktionsprioritetsvärdena (lSVC_L1) och logistisk regressionsklassificerare med de högst rankade L1-funktionsprioritetsvärdena (LR_L1).
Klassificerarpoolen använder sju olika klassificerare för att skapa klassificeringsmodeller. Dessa klassificerare består av linjär stödvektormaskin (SVC), Gaussisk Naïve Bayes (GNB), logistisk regressionsklassificerare (LR), k-närmaste granne, med k inställt på 5 som standard (KNN), XGBoost, slumpmässig skog (RF) och beslutsträd (DT).
Den slumpmässiga uppdelningen av datauppsättningen i train: testdelmängder kan anges på kommandoraden. I det demonstrerade exemplet används förhållandet mellan train: test = 8: 2.
