Term human placentas from lean (pre-pregnancy body mass index (BMI) <25) mothers with uncomplicated pregnancies carrying female offspring were collected and sampled within 15 minutes of delivery by cesarean section (no labor). The placentas were examined for the absence of calcifications and typical development: weighing between 300-600 g with the umbilical cord and membranes removed, round in shape, between 15 – 25 cm in diameter, and umbilical cord inserted into the middle of the placenta. Villous tissue was dissected away from the basal and chorionic plates in 2-3 samples from across the placenta (Figure 1), yielding approximately 100 g of villous tissue as starting material for primary trophoblast isolation. Within 20 minutes of sampling villous tissue, the procedure to isolate primary trophoblasts was started as described here, yielding between 0.8 – 1 x 108 viable cells. The cells were seeded in 6-well culture plates at a density of 3 x 106 (3.3 x 105 cells per cm2). After 24 h of culture, the cells were examined under a microscope for attachment and proper trophoblast morphology was confirmed (individual round cells). The culture media was replaced with complete media containing a series of concentrations of TNFα between 125-104 pg/mL so that at least two wells were included per concentration and vehicle control (complete media only).
Twenty-four hours following TNFα-treatment (48 h of culture), all TNFα medias were replaced with complete media. No appreciable cell death was observed due to treatment with TNFα at concentrations at or below 103 pg/mL. Treatment with 104 pg/mL TNFα was moderately cytotoxic and the cytotoxic effects of this TNFα concentration did not persist after the media was changed as evidenced by Lactate Dehydrogenase (LDH) assays (data not shown). At 72 h of culture, cells were examined for syncytialization under a microscope. Immunocytochemistry for syncytialization and fibroblast contamination revealed relatively pure isolation of trophoblasts (Figure 3). Cellular lysates were harvested according to the protocol described here, yielding between 3-8 µg/µL of total protein per preparation as determined by a BCA assay (data not shown). Western blot analysis in cell lysates from female trophoblasts treated with TNFα showed an upregulation of Rubicon expression in response to concentrations of TNFα up to 250 pg/mL and subsequent downregulation of Rubicon expression at TNFα concentrations greater than 250 pg/mL (Figure 4A and B, 104 pg/mL excluded from analysis based on cytotoxic effects). Likewise, Rubicon is significantly upregulated in flash-frozen villous tissue biopsies from placentas from obese pregnancies with female fetuses compared to lean controls as evidenced by Western blot analysis (Figure 4C and D, n = 6 placentas per BMI class, ANOVA, P<0.05).
| Concentration (%) | 90% DGM (ml) | 1x HBSS (ml) | Layer Thickness (ml) |
| 4x gradient | 4x gradient | Total 34.5 ml | |
| 70 | 14 | 4 | 4.5 |
| 60 | 8 | 4 | 3 |
| 55 | 7.33 | 4.67 | 3 |
| 50 | 3.34 | 2.67 | 3 |
| 45 | 6 | 6 | 3 (13.5 ml mark) |
| 40 | 5.33 | 6.67 | 3 |
| 35 | 4.67 | 7.33 | 3 (19.5 ml mark) |
| 30 | 8 | 16 | 6 |
| 20 | 2.67 | 9.33 | 3 |
| 10 | 1.33 | 10.67 | 3 |
Table 1. Specifications for Making Density Gradients for Density Centrifugation of Primary Trophoblasts.
From left to right, column one specifies density gradient media (DGM, see table of the essential supplies, reagents, and equipment, supplementary material) concentration expressed as percentages of DGM in HBSS. Column two specifies the volume of DGM while column three specifies the volume of HBSS required for making the appropriate percentage of DGM solution. Column four specifies the volume to be added to the 50 mL conical tube to build the gradient, beginning with the most dense layer.
| Trypsin | HBSS | DNase | ||
| Digestion | (total activity; BAEE units) | volume (ml) | (total activity; Kunits) | Total Volume /digestion |
| 1 | 619037 (23.01 ml) | 141.76 ml | 62594 (0.230 ml) | 165 ml |
| 2 | 412691 (15.34 ml) | 94.51 ml | 41729 (0.154 ml) | 110 ml |
| 3 | 313270 (11.65 ml) | 71.74 ml | 31676 (0.116 ml) | 83.5 ml |
| Total | 1345000 (50 ml) | 308 ml | 136000 (0.5 ml) | 358.5 ml |
Table 2. Specifications for the Preparation of Digestion Solution for Primary Trophoblast Isolation Based on the Specific Activity of DNase and Trypsin.
From left to right, the first column specifies the number of the digestions, the second column specifies the trypsin activity required per digestion, the third column specifies the total volume of supplemented HBSS to be added for the appropriate digestion, the fourth column specifies the DNase activity required per digestion, and the final column specifies the volume of digestion solution to be added to the placental tissue for the appropriate digestion.
| HBSS (supplemented with Ca+2 and Mg+2) | Sample Buffer |
| 10% 10x HBSS | 90% 4X Laemmli dye |
| 1.26 mM CaCl2 (anhyd.) | 10% 2-Mercaptoethanol |
| 0.80 mM MgSO4 (anhyd.) | |
| 20.77 mM HEPES | |
| pH to 7.4 with 10N NaOH Make volume up to 1 L with sterile ddH2O Sterile filter into a sterile bottle |
|
| Complete Media | Running Buffer |
| Remove 11% v/v IMDM | 25 mM Tris Base |
| Add 10% v/v FBS | 190 mM Glycine |
| Add 1% 10,000 U/mL Penicillin/Streptomycin (100 U/mL final) | 0.1% SDS |
| pH to 8.3 | |
| Freezing Media | |
| 90% v/v FBS | Transfer Buffer |
| 10% v/v DMSO | 25 mM Tris |
| 190 mM Glycine | |
| Digestion Buffer | 20% Methanol |
| 50 mL Trypsin (26,900 BAEE units/mL) | pH to 8.3 |
| 0.5 mL DNAse (272,000 K units/mL) | |
| Bring to 358.5 ml in supplemented HBSS | TBS |
| 20 mM Tris | |
| RIPA Buffer | 150 mM NaCl |
| 25 mM Tris-HCl | pH to 7.6 |
| 5 mM EDTA | |
| 150 mM NaCl | TBST |
| 0.1% SDS | TBS with 0.1% Tween 20 |
| 0.5% Sodium deoxycholate | |
| 1% Triton X-100 | |
| 1 tablet of protease/phosphatase inhibitor per 10 ml RIPA Buffer |
Table 3. Solutions Required for Isolation and Culture of Primary Trophoblasts followed by Western Blotting.
| % DGM | ml mark | Cell type |
| 10 | 31.5-34.5 | Debris |
| 20 | 28.5-31.5 | |
| 30 | 22.5-28.5 | |
| 35 | 19.5-22.5 | Trophoblasts |
| 40 | 16.5-19.5 | |
| 45 | 13.5-16.5 | |
| 50 | 10.5-13.5 | Lymphocytes |
| 55 | 7.5-10.5 | |
| 60 | 4.5-7.5 | Red blood cells |
| 70 | Below 4.5 |
Table 4. Sedimentation of Trophoblasts by Density Centrifugation.
From left to right, the first column specifies the percentage of DGM (Table 1), the second column specifies the mL mark where the corresponding percentage of DGM is found on a 50 mL conical tube, and the third column specifies what cell type sediments at the corresponding percentage of DGM and mL mark on a 50 mL conical tube.Trophoblasts sediment between 50- 35% DGM, forming distinct opaque bands. Collecting DGM above or below this range will result in contamination of cellular debris and other cell types such as lymphocytes.
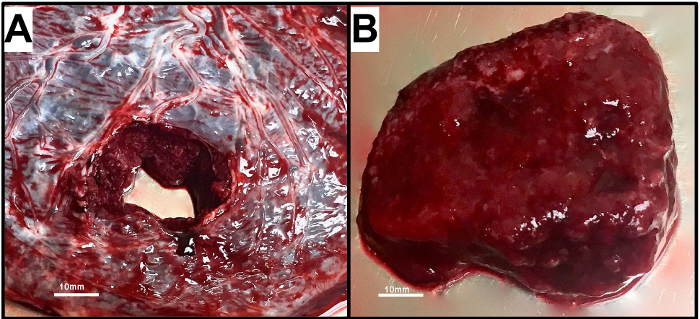
Figure 1. Villous Tissue is Isolated from the Term Human Placenta by Removing the Chorionic and Basal Plates.
A) With the chorionic plate (fetal side) facing upwards, a full thickness sample is excised from the placenta. B) A sample of villous tissue is obtained by removing the chorionic and basal plates. Please click here to view a larger version of this figure.
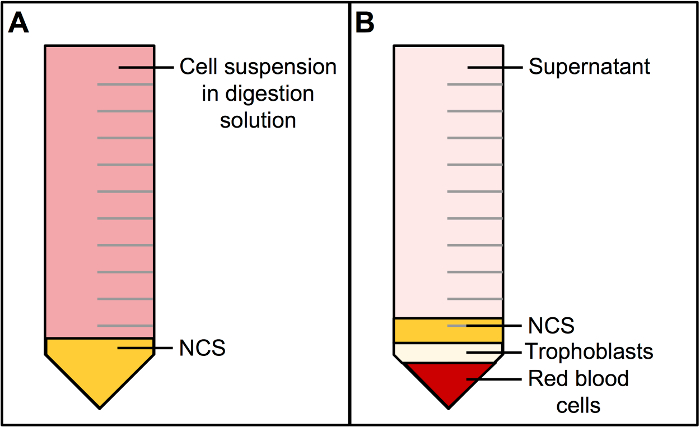
Figure 2. Centrifugation of Cells in Digestion Solution over Newborn Calf Serum results in a Multilayered Cell Pellet.
A) Newborn calf serum (NCS) is layered underneath the cell suspension in digest solution in a 50 mL conical tube. B) Centrifugation of (A) results in a multilayered cell pellet. The bottom-most layer is deep in red color and consists of red blood cells. The layer above includes trophoblasts and is white or beige in color. Above the trophoblast layer is NCS followed by digestion solution (supernatant) to the top of the tube. Please click here to view a larger version of this figure.
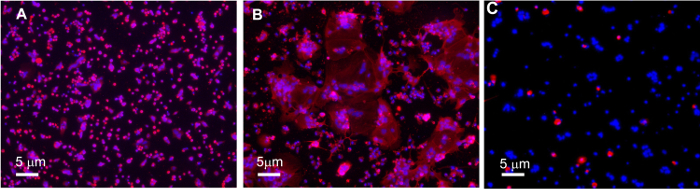
Figure 3. Immunocytological Analysis of Syncytialization and Fibroblast Content in Primary Human Trophoblast Cultures.
A) Representative image of Cytokeratin-7 (red) in cytotrophoblasts after 24 h of culture. B) Representative image of Cytokeratin-7 (red) in syncytiotrophoblasts after 72 h of culture shows multinucleated masses of cells that have fused. C) Representative image of Vimentin (red) in syncytiotrophoblasts after 72 h of culture. Images were acquired on a fluorescent microscope with DAPI (blue) nuclear counterstain. Visualized at 10X magnification. Please click here to view a larger version of this figure.
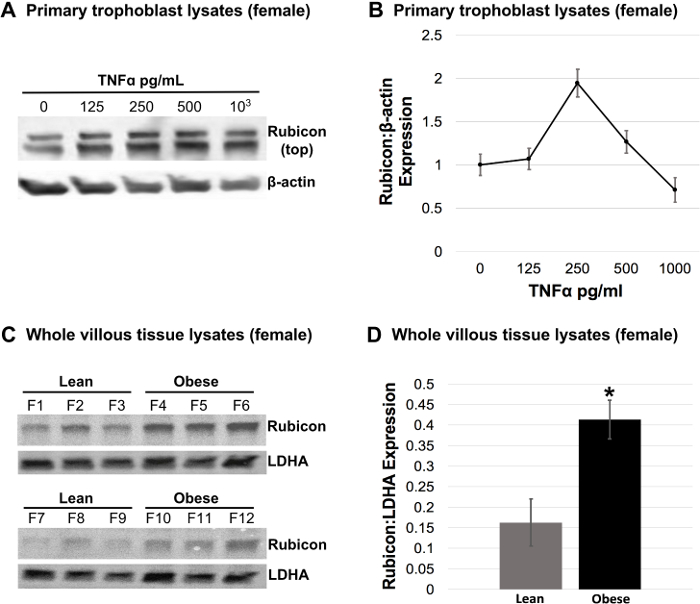
Figure 4. Regulation of Rubicon Expression in Response to TNFα-Treatment in Female Trophoblasts and Endogenous Rubicon Expression in Villous Tissue from Lean Versus Obese Pregnancies with Female Fetuses.
Primary trophoblasts from term placentas from lean mothers with healthy pregnancies carrying a female fetus were isolated and treated with 125, 250, 500, 103, and 104 pg/mL TNFα (or vehicle control). A) Representative Western blot for Rubicon in female trophoblast lysates treated with TNFα. β-actin was used as a loading control. B) Rubicon expression in response to TNFα-treatment in female trophoblasts was quantified from Western blots and normalized to β-actin. Values are mean Rubicon expression per TNFα concentration ± S.E. in n=3 placentas. C) Western blots for Rubicon in whole tissue lysates from flash frozen biopsies of villous tissue from lean versus obese pregnancies with female fetuses (F1 -F12). Lactate dehydrogenase A (LDHA) was used as a loading control. D) Rubicon expression in Western blots from (C) was quantified and normalized to LDHA. Values are mean Rubicon expression per BMI classification ± S.E. in n=6 placentas per BMI class (ANOVA, *P<0.05). Please click here to view a larger version of this figure.