In this section we present representative results for computing the optical absorption spectrum of an aggregate of six YLD 124 molecules, shown in Figure 3a, where the structure of the aggregate was obtained from a coarse-grained Monte Carlo simulation. YLD 124 is a prototypical charge-transfer chromophore that consists of an electron-donating group of diethyl amine with tert-butyldimethylsilyl protecting groups that is connected via a π -conjugated bridge to the electron accepting group 2-(3-cyano-4,5,5-trimethyl-5H-furan-2-ylidene)-malononitrile39. This molecule has a large ground-state dipole moment, ~30 D. Electronic structure calculations for individual molecules were performed using the ωB97X34 functional with the 6-31G* basis set35,36. TD-DFT calculations used the Tamm-Dancoff approximation40. Partial atomic charges were computed with the CHelpG population analysis method37.
The Hamiltonian for this system, constructed using the protocol described in this paper, is shown in Table 1.
The absorption spectrum calculated for this excitonic Hamiltonian is shown in blue in Figure 3b. Because there are six molecules with only a single bright excited state for each molecule, a 6-by-6 excitonic Hamiltonian was generated, resulting in six transitions. The eigenvalues of this Hamiltonian are the lowest six excited state energies for the molecular aggregate. The height of the vertical lines represents the oscillator strength fi for each transition from the ground to the ith excited state of the molecular aggregate. It can be found using the expression29
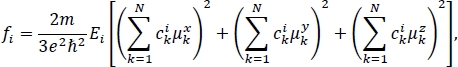
where m is the electron mass, e is the elementary charge, ħ is the reduced Planck’s constant, N is the total number of molecules in the aggregate, Ei is the eigenvalue that corresponds to the ith excited state of the molecular aggregate, cki is the expansion coefficient for the contribution of the kth molecule in the aggregate to the ith excited state of the aggregate written in the basis of bright excited states on individual molecules, and μkα are the components of the transition dipole moment vector for the ground to bright excited state of the kth molecule in the aggregate, α = x, y, z . The values of Ei and cki are found by solving the eigenvalue equation for the Hamiltonian matrix (the time-independent Schrödinger equation). The values of μkα can be found in the “.log2” files that are generated in step 5.2 of the protocol. The total spectrum is a smooth line created by summing over Gaussian functions centered at each of the excitation energies and weighted by the corresponding oscillator strengths29.
For comparison, the spectrum computed from an all-electron TD-DFT calculation on the entire molecular aggregate is shown in magenta. For these spectra, the integrated intensity of the exciton spectrum is larger than the TD-DFT spectrum (Iexc/ITD-DFT = 1.124) and the difference in the mean absorption energies is Eexc – ETD-DFT = 0.094 eV. These offsets are systematic for molecular aggregates of a given size and can be corrected for to obtain very good agreement between exciton model and TD-DFT spectra. For instance, for a set of 25 molecular aggregates that each consists of 6 YLD 124 molecules, the average integrated intensity ratio Iexc/ITD-DFT = 1.126, with a standard deviation of 0.048, and the difference in the mean absorption energies is Eexc – ETD-DFT = 0.057 eV, with a standard deviation of 0.017 eV. The exciton model and TD-DFT spectra shown in Figure 3b also have similar shapes, as characterized by Pearson’s product-moment correlation coefficient41 between them of 0.9818 and Pearson’s product-moment correlation coefficient between their derivatives of 0.9315. On average, for a set of 25 molecular aggregates that each consists of 6 YLD 124 molecules, the agreement in spectral shape is even better than for the example shown, with values of 0.9919 (standard deviation of 0.0090) and 0.9577 (standard deviation of 0.0448) for the two Pearson’s coefficients, respectively29. Our earlier work suggests that the spectral shape is primarily determined by local electrostatic interactions between chromophores in the aggregate that are accounted for in the exciton model described in this paper, whereas the excitation energy and intensity depend considerably on the mutual polarization between the chromophore and its environment that the model neglects29.

Figure 1: An isosurface for the transition density plotted for a single molecule of YLD 124. The the positions of atomic charges on surrounding molecules are shown by gray dots. Please click here to view a larger version of this figure.
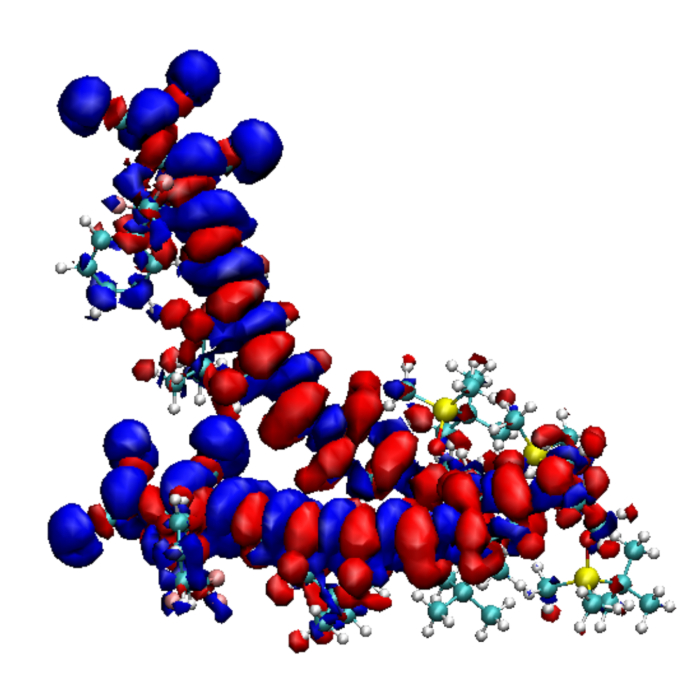
Figure 2: The transition densities plotted for two molecules of YLD 124, i and j, that are used for computing the excitonic coupling bij between these molecules. The surrounding charges are not shown. Please click here to view a larger version of this figure.
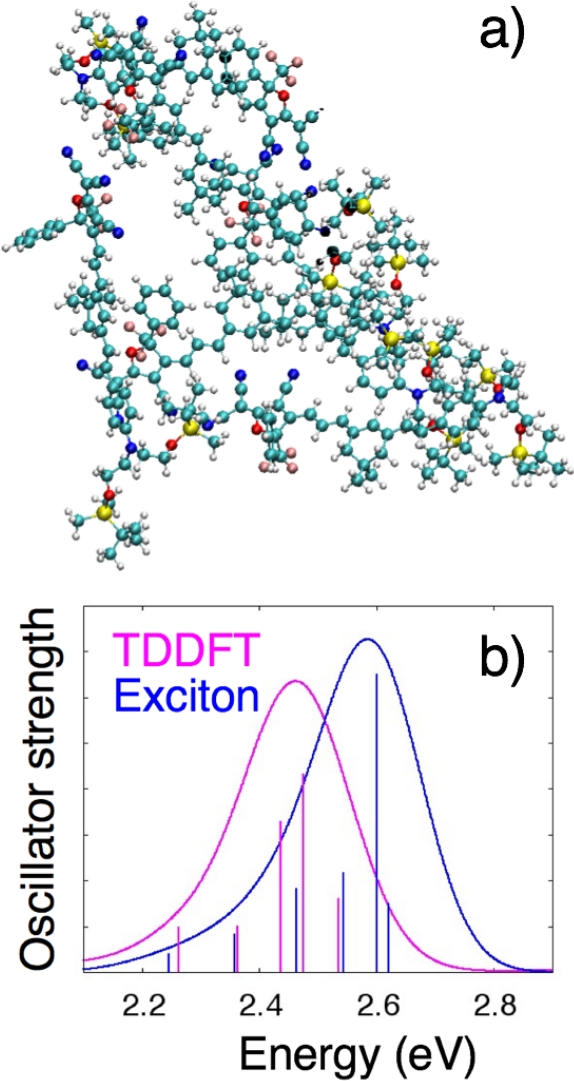
Figure 3: The structure and calculated spectrum for an aggregate of six YLD 124 molecules. (a) The aggregate structure used in the sample calculation. (b) The corresponding absorption spectra created using the exciton model Hamiltonian (blue) and an all-electron TD-DFT calculation on the entire aggregate (magenta). Please click here to view a larger version of this figure.
| 2.4458 | -0.0379 | -0.0899 | 0.0278 | -0.0251 | 0.0120 |
| -0.0379 | 2.4352 | -0.0056 | -0.1688 | -0.0070 | -0.0085 |
| -0.0899 | -0.0056 | 2.5111 | 0.0032 | 0.0239 | 0.0794 |
| 0.0278 | -0.1688 | 0.0032 | 2.3954 | 0.0057 | 0.0073 |
| -0.0251 | -0.0070 | 0.0239 | 0.0057 | 2.5171 | -0.0211 |
| 0.0120 | -0.0085 | 0.0794 | 0.0073 | -0.0211 | 2.5256 |
Table 1: The Hamiltonian for a sample calculation on the aggregate of six YLD 124 molecules shown in Figure 3a. The diagonal elements are the excitation energies of individual molecules; the off-diagonal elements are the excitonic couplings between molecules (all values are in eV).