The effect of CXCL12 stimulation on the intracellular Ca2+ mobilization in U87.CD4.CXCR4+ and U87.CD4 cells was evaluated with the Ca2+ mobilization assay. Instead of 20 µL of test compound that would normally be added during the first pipetting step of the protocol (Figure 1), assay buffer was added to the fluo-2 AM loaded U87.CD4.CXCR4+ cells in the measurement plate. During the second dispensing step, different concentrations of CXCL12 (0.2-102.4 nM, final concentration) were dispensed in the measurement plate. A dose-dependent increase in fluorescence, correlating with a dose-dependent increase of the release of Ca2+, is demonstrated (Figure 2A). Here, the negative control sample represents those wells in which at both additions only assay buffer was added to the measurement plate (i.e., 0 nM CXCL12), resulting in the absence of a response (Figure 2A). Increasing amounts of CXCL12 induce increased levels of fluorescence that decay over time (Figure 2A). From these data, a dose response curve was generated based on the "Max-Min response over baseline" between measurement 84 (i.e., the first measurement after CXCL12 addition) and measurement 243 (i.e., the final measurement in the protocol). Using nonlinear regression, the EC50 value (i.e., the concentration needed to evoke the half maximal response) was determined and corresponds to 1.14 nM (Figure 2B). No fluorescent Ca2+-related response was evoked by CXCL12, even at high concentration (102.4 nM), when U87.CD4 cells lacking functional CXCR4 expression were used. This demonstrates the receptor-specificity of the measurement evoked by CXCL12 (Figure 2C).
A key application for this cell-based assay is the identification of small molecules specifically targeting CXCR4 and capable of inhibiting the CXCL12-induced Ca2+ mobilization. If active compounds ("hits") are identified, they can be further characterized by testing serial dilutions of the compound and analyzing the inhibitory potency in more detail. As an example, the effect of the bicyclam AMD3100, a well-established small molecule CXCR4-specific receptor antagonist with potent anti-HIV-1 activity21,22, is illustrated in Figure 3. A concentration series of AMD3100 (n = 4, 3 µM down to 1.37 nM final concentration in a 1/3 dilution series) was dispensed on U87.CD4.CXCR4+ cells during the first step of the protocol. The compound was then allowed to incubate with the cells for ~ 10 min during which the fluorescence signal was recorded continuously. Then 6.4 nM of CXCL12 (50 ng/mL, final concentration) was added to all wells of the cell plate simultaneously to induce the CXCR4-mediated Ca2+ mobilization. Dose-dependent inhibition of this response by the different dilutions of AMD3100 is shown in Figure 3A. In this case, only the part of the graph following addition of CXCL12 is shown. As a negative control (blue line) only buffer was applied in the assay (no pre-incubation with AMD3100, no CXCL12 added to the wells), resulting in the expected lack of response. The positive control (red line) corresponds to the samples in which 6.4 nM CXCL12 was added to wells without prior incubation of AMD3100 (Figure 3A). Based on these defined negative and positive controls the Z' value in this assay typically is between 0.5 and 1. In order to determine the inhibitory potency of AMD3100 a dose-response curve was generated by non-linear regression resulting into a calculated IC50 value of 770.8 nM (Figure 3B). To further illustrate the behavior of a non-active compound, maraviroc was included in this experiment. Maraviroc is a small molecule specifically inhibiting the CC chemokine receptor 5 (CCR5) thereby blocking (CCR5)-tropic HIV-1 infection23 . Even at high concentration (10 µM final concentration) no inhibitory effect of this compound was observed on the CXCR4-mediated Ca2+ response (Figure 3A).
Finally, to demonstrate that this Ca2+ mobilization assay can also be applied to study other GPCRs, a serial dilution of the CC chemokine ligand 5 (CCL5), the endogenous ligand for CCR5, was added to cells expressing CCR5 (U87.CD4.CCR5+) using exactly the same experimental conditions and hardware settings. A dose-dependent fluorescent Ca2+ response is demonstrated after addition of a serial dilution of CCL5 (Figure 3C). In addition, and in contrast to its effect on CXCR4, pre-incubation with 100 nM (final concentration) of maraviroc strongly inhibited this response (Figure 3D).
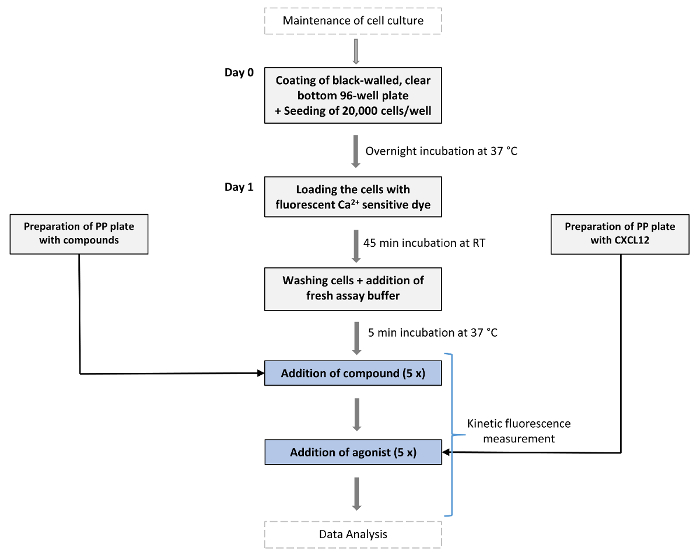
Figure 1: Schematic overview of the assay's workflow. On day 0 cells expressing the GPCR of interest (in this case CXCR4) are seeded in black-walled 96-well plates with clear bottom and are grown overnight at 37 °C and 5% CO2. At day 1 the fluorescence-based Ca2+ assay is performed. Cells are first loaded with a fluorescent Ca2+-sensitive dye (fluo-2 AM) and incubated for 45 min at RT in the dark. A "chemokine plate" and "compound plate" are prepared (PP = Polypropylene). After incubation with loading dye, seeded cells are washed once with assay buffer (150 µL/well) after which 80 µL/well of assay buffer is added. All plates are then incubated for 5 min in the device at 37 °C before starting the assay. Then, compounds of interest (e.g., small molecules) are dispensed at the desired concentration into the wells of the measurement plate and allowed to incubate for ~ 10 min while fluorescence is continuously measured. Next, a fixed concentration of the endogenous agonist of the GPCR (here CXCL12) is added to evoke Ca2+ release and fluorescence is further recorded over time. Please click here to view a larger version of this figure.
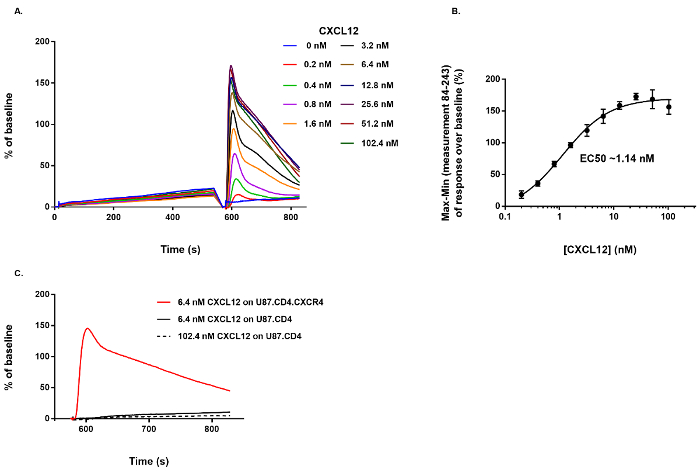
Figure 2: Agonist activity of CXCL12 on CXCR4+ cells and specificity of the detected response. (A) The dose-dependent effect of CXCL12 on the Ca2+ mobilization in U87.CD4.CXCR4+ cells. (B) Based on the Max-Min response over baseline between measurement 84 and 243 a dose-response curve was generated and the EC50 value calculated (n = 4, mean ± SD). (C) No response was induced by CXCL12 when it was dispensed on U87.CD4 cells lacking CXCR4. Please click here to view a larger version of this figure.
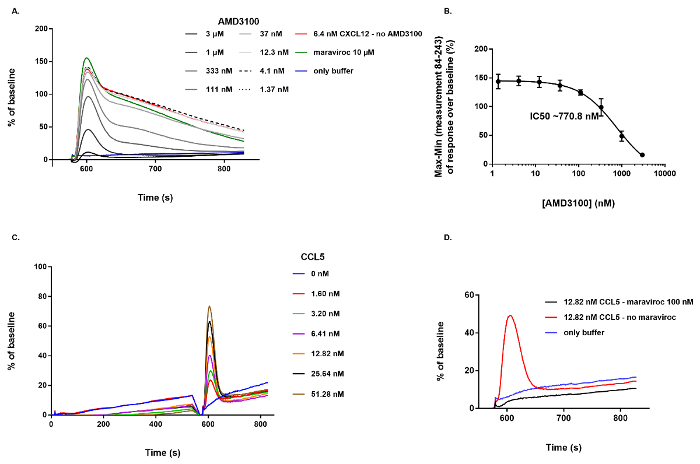
Figure 3: Illustration of the effect of a CXCR4 antagonist (AMD3100) and a non-active compound (maraviroc) on CXCR4+ cells and activation of CCR5 by its endogenous agonist, CCL5. (A) Dose-dependent inhibitory effect of AMD3100 on the Ca2+ mobilization evoked by adding 6.4 nM CXCL12 to U87.CD4.CXCR4+ cells. Maraviroc (at 10 µM final concentration) showed no inhibitory effect on the CXCL12-induced Ca2+ response. (B) Based on the Max-Min response over baseline between measurement 84 and 243, an inhibitory dose-response curve was generated and the IC50 value was calculated to be 770.8 nM (n = 4, mean ± SD). (C) Dose-dependent activation of CCR5 by its endogenous agonist, CCL5. (D) Inhibition of the CCR5-mediated Ca2+ response by pre-incubation of the cells with 100 nM of maraviroc. Please click here to view a larger version of this figure.
