The protocol was originally adapted from B. Ren, 2001. It represents a slight modification of the protocol by Odom et al.5 that can be found at http://jura.wi.mit.edu/cgi-bin/young_public/navframe.cgi?s=22&f=appendices_downloads
1. Pre-block and binding of antibody to magnetic beads (Should be performed the night before next step)
- Wash 100 μL of Dynabeads Protein G (per IP, combine for multiple IPs) in 1 mL of fresh BSA/PBS solution (50 mg BSA in 10 mL PBS- This solution will last for one week).
- Collect the beads using a magnetic stand and repeat the washing procedure two more times.
- Next, add 10 μg of antibody to 250 μl of the beads slurry in PBS/BSA solution (per IP) and incubate overnight on a rotating platform at 4°C.
- Upon completion, wash the beads three times in 1 ml of the PBS/BSA solution and then resuspend in 10 μl of the PBS/BSA solution (per IP).
2. Cell cross-linking
- To begin this procedure, grow approximately 108 cells for each immunoprecipitation, or IP. Here, diffuse histiocytic lymphoma U937 cells are used and induced for monocytic differentiation with TPA for 96 hours.
- Following cell growth, add formaldehyde solution directly to the media to a final concentration of 1%. Then, swirl the flasks briefly and allow them to sit at room temperature for 10 minutes. Then, aspirate the media and rinse the cells with 15 mL of ice-cold PBS. Repeat this wash once.
- Next, add 6 mL of Lysis Buffer 1 (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, containing protease inhibitors) to each of the flasks on ice. Then, rock the flasks for 20 minutes at 4°C.
- Finally, harvest the cells using a cell scraper and transfer them to 15 mL conical tubes. At this point the cells can be stored at -80°C.
3. Cell Sonication
- If the cells were frozen, thaw them out. Once thawed, spin the cells down at 3,000 rpm for 10 minutes at 4°C and discard the supernatant. Then, resuspend the cells in 6 mL of Lysis Buffer 2 (10 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1mM EDTA, 0.5 mM EGTA, containing protease inhibitors). Rock the tubes gently at room temperature for 10 minutes.
- Repeat centrifugation and resuspend the cells in 2.5 mL of Lysis Buffer 3 (10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine, containing protease inhibitors).
- Next, prepare the suspension for sonication by placing it in a beaker of icy water with the microtip of a Branson 450 Sonifier set to between 50 and 60% amplitude.
- Sonicate the solution for a 30 second constant burst and cool on ice for 1 minute. Repeat these pulses 10 to 15 times. Then, pipette the contents of the tube up and down and transfer them to a new tube. Continue to pulse and cool the solution an additional 5 times.
- Following sonication, add 10% Triton X-100 to 1/10 of the solution volume. Transfer the lysates to 1.5 mL centrifuge tubes, and spin the debris out in a microcentrifuge. Then transfer the cell lysate to a new tube.
4. Chromatin Immunoprecipitation
- Prior to chromatin immunoprecipitation, or ChIP, save 50 μL of the cell lysate as the input sample. Then, combine the cleared cell lysate with Dynabeads pre-bound to antibody that have been previously prepared as described above. Rock the mixture, in addition to the 50 μL input sample, at 4°C overnight.
- Wash the beads with 1 mL of the Wash Buffer (50 mM HEPES-KOH, pH 7.6, 0.5 M LiCl, 1mM EDTA, 0.7% sodium deoxycholate, 1% NP-40). Then, use a magnetic stand to collect the beads and remove the supernatant. Repeat this wash 6 to 8 times.
- Perform a final wash with 1 mL TE-plus-50 mM NaCl (10 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA). Spin the beads in a microcentrifuge at 3,000 rpm for 2 minutes at 4°C. Following centrifugation, aspirate any residual TE buffer.
- Then, add 100 μL of Elution Buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 1% SDS) to the samples. Immediately after, incubate the samples at 65 °C for 10 to 15 minutes. Scratch the tubes against a 1.5 mL tube rack every 2 minutes to keep the beads in the suspension.
- Collect the beads using centrifugation and the magnetic stand, and transfer the supernatant to a PCR tube. Then, retrieve the previously saved input sample and add 3 volumes of the Elution Buffer.
- Finally, place the IP and input samples into the thermal cycler overnight at 65 °C to reverse the cross-linkages.
- The following day, add 1 volume of TE buffer to the samples. Then, add RNase A to a final concentration of 0.2 μg/μL. Incubate the samples in the thermal cycler for 1 to 2 hours at 37 °C.
- After incubation, add proteinase K to a final concentration of 0.2 μg/μL. Then, incubate samples in the thermal cycler at 55 °C for 2 hours.
- Next, extract the samples once with one volume of phenol. Extract the samples a second time with one volume of phenol:chloroform:isoamyl alcohol. Finally, extract the samples once more with one volume of chloroform:isoamyl alcohol.
- Then, add 30 μg of glycogen to each sample. Also, add NaCl to a final concentration of 0.2 Molar and two volumes of ethanol to the samples. Incubate them for 30 minutes at -80 °C. After incubation, spin the samples and decant the supernatant. Wash the pellets with 500 μL of 75% ethanol. Then, dry the pellets and re-suspend them in 40 μL of water.
- To check the sizes of the DNA fragments produced by the sonication procedure, load 5 μg of the purified input sample in a 1.8% agarose gel and run the gel. The sonication procedure should produce fragments in the range of 150-350 base pairs. However, if the fragments do not fall in this range, the sonication procedure should be adjusted by varying the number of bursts and the amplitude.
- To check robustness and specificity of ChIP, the enrichment in control binding regions by performing gene-specific PCR with 1 μl of the IP samples and a dilution series of the input sample. Two to three “bound” regions and an unbound control region need to be selected in order to test quality of IP and input samples.
5. Amplification of genomic DNA
- Starting with 34 μl of the IP sample or 200 ng of the input sample, perform end repair, ‘A’ tail addition and ligation of adapters to DNA fragments using Genomic DNA Sample Prep Kit http://www.illumina.com/systems/genome_analyzer.ilmn (Illumina, Inc., San Diego, CA).
- Purify the DNA using a MinElute PCR Purification Kit and then elute it in buffer EB (10 mM Tris Cl, pH 8.5) that has been pre-warmed to 50 °C. For example, use 23 μL and 46 μL for the final elution of IP and input DNA samples, respectively.
- Make the PCR reaction using 23 μL of DNA with adapters, and 25 μL Phusion DNA polymerase from the kit, 1 μL of 20 μM Sol_PCR_1, and 1 μL of 20 μM Sol_PCR_2. Run the PCR reaction as follows: 1) 30 seconds at 98°C; 2) 10 seconds at 98°C; 3) 30 seconds at 65°C; 4) 30 seconds at 72°C; repeating steps step 2-4 18 times and then 5 minutes at 72°C, finally holding at 4°C.
- Following PCR amplification, purify the DNA with the QIAGEN MinElute PCR Purification Kit. Pre-warm 15 μL of buffer EB to 50 °C and elute the DNA. Dilute 0.5 μL of the sample 1:4 and perform a Nanodrop reading.
6. Gel purification of amplified products
- To purify the amplified products, prepare a 1.8% agarose gel of 50 mL using HPLC-grade water. Add TAE and ethidium bromide after the agarose has melted. Then, pour the gel. Load the gel after adding 4 μL of loading buffer to the input and IP samples. Then, run the gel at 120 volts for 45 minutes.
- Analyze the gel after it has run. The gel should reveal that fragments in the range between 150 and 350 base pairs are produced. They represent genomic DNA fragments between 50 and 250 base pairs in length and an adapter.
- Excise the region of gel containing the material in the 150 350 base pair range with a scalpel. Take care to avoid excising the adapter-adapter band, which runs at about 120 base pairs. Recover the DNA using a QIAquick Gel Extraction Kit per manufacturer’s instructions. Specifically, use one column from the QIAquick Gel Extraction Kit for a gel slice of 400 mg or less. To begin the extraction process, add 3 volumes of QG buffer to 1 volume of gel. Add 1 volume of isopropanol and load the mixture on the column. Use 0.5 ml of QG to wash the column, followed by standard procedure described in kit manual. Use H2O pre-warmed to 50°C to elute the DNA. Incubate the column with 30 μl of H2O for 5 minutes at 37°C before spinning it down.
- Dry the sample down to precisely 11 μL in a Speedvac without heat. Remove 1 μL of the sample and measure the DNA concentration using a Nanodrop spectrophotometer.
- Finally, identify enriched gene products using the Illumina Genome AnalyzerIIe as described at http://genesdev.cshlp.org/content/suppl/2008/12/15/22.24.3403.DC1/GuentherSuppMat.pdf 6.
7. Representative Results
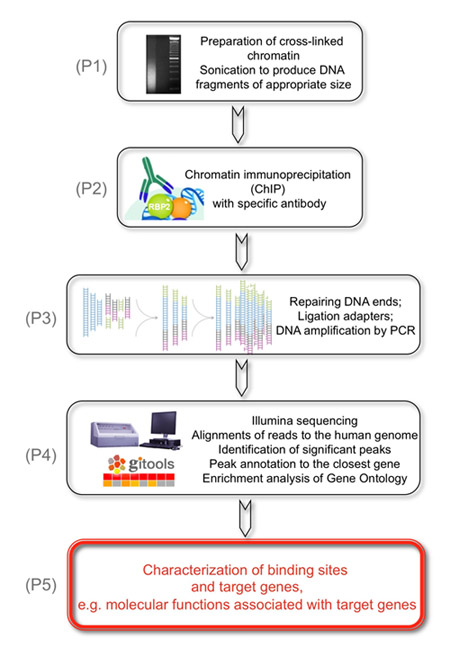
Figure 1. The overall goal of the following experiment is to identify genomic targets of KDM5A/JARID1A/RBP2 histone demethylase.
(P1) This is achieved by preparation of cross-linked chromatin that is sonicated to produce DNA fragments of appropriate size. (P2) As a second step, RBP2 bound DNA fragments are immunoprecipitated with RBP2 antibodies. (P3) Next, the recovered DNA fragments are repaired at the ends and adapters are ligated onto the ends of genomic DNA to prepare libraries of genomic DNA for analysis on the flow cells in the Illumina Cluster Station. The DNA libraries are amplified by PCR using low number of cycles. (P4) Finally, illumina sequenced short reads are uniquely aligned to the human genome, significant peaks are identified and annotated to the closest genes in order to identify RBP2 enriched regions. (P5) Results are obtained that show molecular functions associated with RBP2 target genes based on genome-wide identification of RBP2 enriched regions.
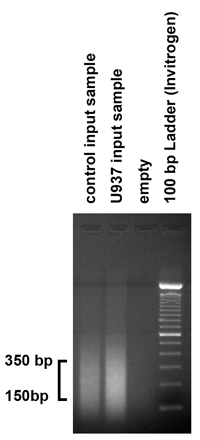
Figure 2. Checking the results of chromatin sonication To check the sizes of the DNA fragments produced by the sonication procedure, 5 μg (1/10) of the purified input sample was loaded on 1.8% agarose gel. As expected, our sonication procedure produced fragments in the range of 150-350 bp. However, if the fragments do not fall in this range, the sonication procedure should be adjusted accordingly, by varying the number of bursts and the amplitude.
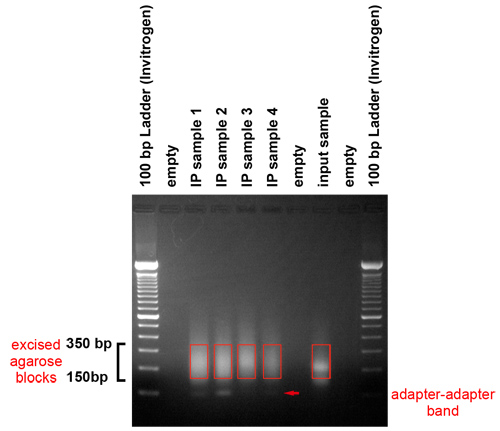
Figure 3. Gel purification of amplified products. The PCR products are run on a gel to remove the adapters and select a size-range of templates for the cluster generation platform. This gel shows that fragments in the range between 150 and 350 base pairs were produced. They represent genomic DNA fragments between 50 and 250 base pairs in length and an adapter. We excise the selected region of gel with a scalpel. Care should be taken to avoid the adapter-adapter band, which runs at about 120 base pairs.
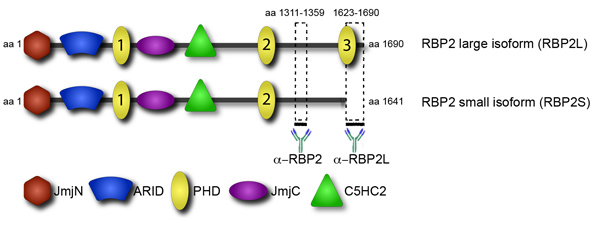
Figure 4. Isoform-specific antibody allows distinguishing between large and small isoforms of RBP2. RBP2 protein structure is presented in domain view. RBP2 contains several domains: the catalytic histone demethylation JmjC domain and associated JmjN domain, an ARID domain capable of sequence-specific DNA binding, a C5HC2 zinc finger that may potentially interact with DNA or other proteins, and multiple PHD domains. The two anti-RBP2 antibodies that allow distinguishing between RBP2 isoforms were derived against the RBP2 fragments indicated by thick lines.
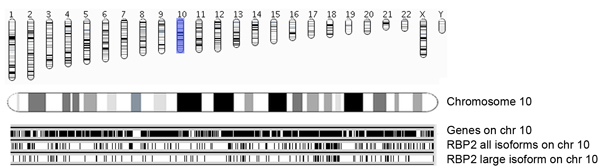
Figure 5a. Overview of RBP2 binding regions along with chromosome and Ensembl genes. Genomic coordinates of identified enriched RBP2 (all isoforms and large isoform) binding regions are presented chromosome wise. Occupances of Ensembl genes (version 54; hg18) in the chromosomes (chromosome 10 in this picture) are presented as black bars (top panel). Each RBP2 binding region is represented as a vertical line, where the middle panel shows all RBP2 isoforms binding regions on chromosome 10, and the bottom panel shows RBP2 large isoform binding regions. The middle and bottom panels give overview of overlapping and specific occupancy of RBP2 isoforms on chromosome 10.
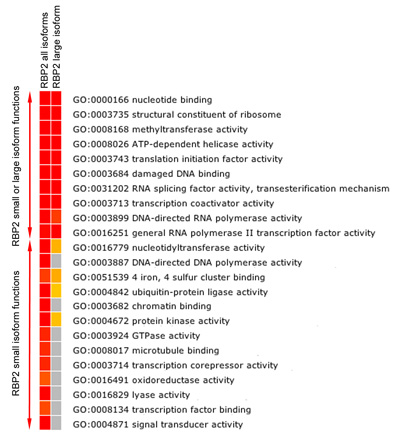
Figure 5b. Functional enrichment analysis of RBP2 target genes. Heatmap showing FDR corrected significantly (p-value ≤0.05) enriched GO Molecular Function categories among the genes bound (closest genes to the RBP2 binding region) by RBP2 (all and large isoforms). Colors toward red indicate high statistic significance, yellow indicates low statistic significance, and gray indicates no statistic significance. Enrichment analysis shows the overlapping and isoform specific molecular functions of RBP2 target genes.
