Note: In vivo crystallization has been reported in many organisms including in bacteria, yeast, plants, insects and mammals (reviewed in reference 21). Crystallization of recombinant proteins has also been achieved in the laboratory using transient transfection of mammalian cells and baculovirus infection of insect cells. The following protocol has been developed using the Bombyx mori cypovirus 1 (BmCPV1) polyhedrin gene cloned in a recombinant baculovirus under the baculovirus polyhedrin promoter, generated as per instructions in reference 22. Thus, although this protocol may be adapted to other cell types (e.g. mammalian cells), we describe here the procedures for insect cells. The protocol assumes that over-expression of the protein of interest has already been achieved. Methods for the production of a recombinant baculovirus and its use for expression of the protein of interest are available in references 23,24,25,26,27.
1. Identification of Crystal-containing Cells
- If cells are grown in monolayer, directly inspect them in the flask. If cells are grown in liquid culture, use the following protocol.
- Using a sterile serological pipette, transfer 500 µL of cells into a microcentrifuge tube. Take care to ensure that the sterility of the main culture is maintained; handle the cells in a Class II biosafety cabinet and follow standard aseptic techniques.
- Pipet 5 µL of cells from the microcentrifuge tube onto a glass slide.
- Carefully place a glass coverslip onto the liquid, avoiding the formation of air bubbles. If the slide cannot be visualized within 15 min, seal the coverslip with vacuum grease to avoid evaporation.
- Image the flask or slide with an inverted microscope. If available, use phase contrast and/or differential interference contrast microscopy (DIC). Both techniques enhance the contrast in transparent samples and emphasize lines and edges, thus facilitating the identification of in vivo crystals.
- Carefully examine the cells. Start at 200X magnification and zoom in at maximal magnification when detecting a potential crystal. The presence of crystals may be suggested indirectly by changes in the cell morphology (Figure 2). Look for sharp edges and changes in refractivity (if using phase contrast or DIC). Known in vivo crystals adopt different shapes including rods, stacks of needles, cubes, pyramids and polyhedra (see Figure 2 and Table 1 for some examples).
Note: The proportion of cells containing crystals will be different in each case and can even vary between preparations but in general one should not expect to find crystals in all cells. Similarly, the number of crystals per cell is also variable (see Table 1), and more than one crystal may be found in one cell. These factors need to be optimized where possible by adjusting the multiplicity of infection, altering the length of expression and varying the protein construct. On one hand, conditions of protein expression and cell growth that maximize the number of crystal-containing cells will improve productivity (e.g. higher multiplicity of infection and late harvesting of cell culture). On the other hand, conditions where cells contain a single crystal facilitate data collection (e.g. low multiplicity of infection). Given the enrichment brought by flow sorting described in step 2.2, we recommend aiming for larger crystals (e.g. a single crystal per cell) even if the proportion of crystal-containing cells is low. - If crystals are identified, record their position (when imaging monolayer in a flask), and estimate the percentage of crystal-containing cells. If available, use a microscope equipped with a camera capable of tracking changes at a single cell level.
- For cells in suspension, determine the degree of cell viability following the protocol detailed in reference 28. For cells cultured in a monolayer, estimate by visual inspection the approximate degree of confluence and amount of detached cells.
- Monitor the crystal growth, cell growth and viability twice a day.
- Harvest the cells as soon as crystal growth appears to stop. For remarkably robust crystals like viral polyhedra, extended incubation times (>4 days) increase the number of large crystals. However, for typical protein crystals, we recommend to harvest cells when viability drops below 80% to prevent damage caused by cell lysis.
- Remove the flask from the incubator and proceed to step 2.2 as soon as possible. Keep the cells on ice during the following steps, unless specified otherwise.
2. Sample Purification
Note: We describe two methods of analysis of the in vivo crystals from purified crystals and in cellulo, respectively.
- Purification of crystals
Note: Unless there is evidence that the crystals of interest are sensitive to low temperature, work on ice and use ice-cold buffers to avoid crystal degradation.- Pipet 50 mL of infected Sf9 cells in a 50 mL conical centrifuge tube. This represents ~4 x 108 cells in a typical experiment.
- Pellet cells at 450 x g for 10 min at 4 °C.
- Discard the supernatant and resuspend the pellet in 40 mL of chilled phosphate buffered saline (PBS), pH 7.4.
Note: PBS is proposed as a starting point for purification as a standard isotonic buffer with the aim of mimicking the cellular environment where the in vivo crystals grew. It has been used for most in vivo crystals grown in cell culture to date9,14,29. Alternatively, DMEM media has also been successfully used in the purification of in vivo crystals from mammalian cells 19. If crystals visibly suffer from their transfer in PBS or present poor diffraction, this parameter should be investigated as a potential source of degradation. - Sonicate the resuspended pellet for 30 s at 10 mA using a sonicator equipped with a 19-mm probe. To avoid heating the sample, place it in a beaker filled with ice while sonicating. Chemical or mechanical cell disruption may be used as alternatives if crystals appear to be damaged by sonication as assessed in step 2.1.7.
- Centrifuge at 450 x g for 10 min at 4 °C. After centrifugation, 2 layers are apparent: an upper layer of cellular debris, which is pale brown, and a lower pellet of polyhedrin crystals, which is white and chalky.
- Discard the supernatant and remove the upper layer by cautious pipetting. Resupend the lower pellet in 40 mL of chilled PBS.
- Assess the quality of the samples by imaging with a light microscope.
- Pipet 5 µL of cells or purified crystals onto a glass slide. This can be done at room temperature.
- Carefully place a glass coverslip onto the liquid, avoiding the formation of air bubbles.
- Image the slide with an inverted microscope at 200X magnification. Image the sample within 15 min of its preparation on the slide. If the slide cannot be visualized within 15 min, seal the coverslip with vacuum grease to avoid evaporation.
NOTE: In the case of polyhedra, crystals will appear as refringent cubes of ~1 – 10 µm per side. Check the integrity of the crystals looking for signs of loss of sharpness (e.g. round edges), cracks or dissolution. Also monitor the presence of cell debris that will appear as clumps and objects of irregular size and shape.
- Repeat steps 2.1.5 to 2.1.7, halving the volume of PBS used to resuspend the pellet at each cycle until the crystals appear free of debris. Resuspend the final pellet in 1 mL of chilled PBS and transfer the crystals to a microcentrifuge tube. Store the purified crystals at 4 °C.
- Estimate the concentration of crystals by using a hemocytometer as described by the manufacturer. Count crystals as if counting cells.

Figure 3: Purified In Vivo Microcrystals. Representative purification of crystals of the BmCPV1 polyhedrin from Sf9 insect cells showing (a) pelleted cell debris and crystals after sonication, and (b) close-up on the debris and crystal layers. Debris layer can be carefully resuspended in the supernatant and discarded for faster crystal purification. (c) Optical microscopy image of purified polyhedra. Scale bar = 50 µm. Please click here to view a larger version of this figure.
- Isolation of crystal-containing cells by flow-cytometry
Note: To facilitate the definition of gates in flow cytometry, it is recommended to grow a control culture in parallel to the one expressing the protein of interest. This can be done by infecting a flask of Sf9 cells with a non-relevant recombinant baculovirus, or by preparing mock-infected cells. Sorting and imaging of samples is usually done at room temperature.
CAUTION: Propidium iodide is a suspected carcinogen and should be handled with care.- Pipet 4 mL of infected Sf9 cells – representing ~3 x 107 cells – in a tube adapted to flow-cytometry analysis. Also, prepare a second tube with an equivalent number of Sf9 cells infected with a non-recombinant baculovirus.
- Add propidium iodide (PI) to both samples at a final concentration of 1 µg/mL just prior to flow cytometric evaluation. This step is necessary to identify dead cells, cell clumps and debris that may alter or mask cell distribution on the forward- and side-scattering plot. Prepare PI stocks of 1 mg/mL in water and store at 4 °C protected from light.
- Apply cells to a standard flow cytometer capable of cell sorting according to forward- (FSC) and side-scattering (SSC). Analyze at least 20,000 events of the control (i.e. non-crystal containing) sample. After discarding dead cells, cell clumps and debris from the analysis, generate the FSC to SSC plot.
- Analyze at least 20,000 events of the crystal-containing sample. Compare the FSC to SSC plot with the mock one. Look for the appearance of a distinct population corresponding to crystal-containing cells. Typically, cells with intracellular crystals will have a higher SSC due to an increased in morphological complexity. The FSC may also increase if crystal growth causes an increase in the volume of the cell. Define the gates of the flow sorter to isolate cell populations corresponding to the areas of the scatter plot that differ from the mock plot. Image the sorted samples by light microscopy as described in step 1 to confirm which population is associated with crystal-containing cells.
- Using the gates defined in step 2.2.4, sort crystal-containing cells in PBS, or in any other isotonic buffer. Record the number of crystal-containing cells sorted as well as the final volume to facilitate subsequent steps. Store the sorted cells on ice or at 4 °C.
Note: In an hour, >300,000 polyhedra-containing cells are sorted on a cell sorter with a 100 mm nozzle (138 kPa with a frequency of 39 kHz). This amount of cells is enough to prepare >1,000 meshes. - Following the protocol detailed in step 2.1.7, image the cells with a light microscope to confirm the enrichment in crystal-containing cells. Sample preparation for data collection should be done as soon as possible after cell sorting since crystals may degrade over time or be released due to cell lysis.
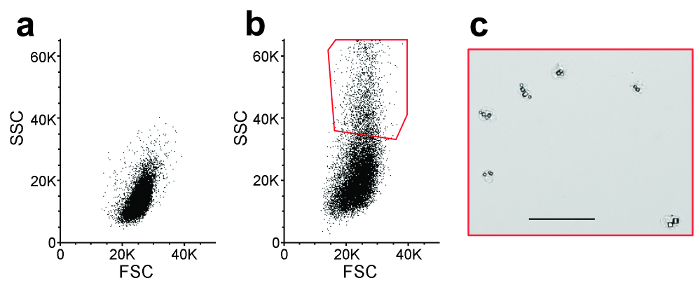
Figure 4: Cell-sorting. Representative flow cytometry scatter plots from (a) non-infected cells (mock), which do not contain crystals, and (b) Sf9 cells infected by a recombinant baculovirus over-expressing for the BmCPV1 polyhedrin. Differences in the scattering pattern between the two populations allowed gating of the crystal-containing cell population (red gate, high SSC values). Each plot represents 20,000 sorting events. SSC: side light scattering; FSC: forward light scattering. (c) Optical microscopy image of a representative population of the sorted cells, showing the presence of intracellular crystals. Scale = 50 µm. Please click here to view a larger version of this figure.
3. Sample Preparation for Data Collection
Note: If cells or crystals are to be derivatized for experimental phasing follow protocol detailed in section 3.1. Otherwise, go directly to sections 3.2 for purified crystals or 3.3 for in cellulo analysis.
- Derivatization of purified or in cellulo microcrystals
CAUTION: Consult all relevant material safety data sheets (MSDS) before use. Most of the chemicals used for experimental phasing are acutely toxic. Use all appropriate safety practices including the use of a chemical fume hood and personal protective equipment. Dispose of waste products in accordance with your institution policy.
Note: The nature of heavy atoms, their concentration and incubation time are given on an indicative basis for crystals of the BmCPV1 polyhedrin. These parameters must be determined experimentally for each new target. Subsequent staining with trypan blue in step 3.3.2 can be omitted if cells are readily colored by the chemical compound used for phasing.- Prepare saturated solutions of the desired heavy atoms (or solutions of other chemicals used for experimental phasing). Typically, volumes lower than 100 µL will be required for each heavy atom. Remove undissolved salts and debris by centrifugation at maximum speed in a microcentrifuge for 5 min at room temperature.
- Prepare aliquots of ~50,000 crystals or sorted cells per heavy atom compound. If PBS is not compatible with the chemical used for derivatization (i.e. precipitate forms and the solution becomes cloudy), pellet the crystals at 4,000 x g for 3 min at room temperature in a microcentrifuge, or the cells at 150 x g for 3 min, remove the supernatant and gently resuspend the crystals or cells in 20 µL of a suitable buffer. Repeat pelleting and resuspension to ensure that no traces of PBS remain. Adjust the final volume to 20 µL for all aliquots.
- Add 20 µL of heavy atom solution or other chemical compound of choice to the crystals or cells. The optimal concentration of heavy atom will depend on a combination of cellular parameters (permeability, off-target proteins …) and the nature of the crystal of the protein of interest. As a starting point, test 3 different concentrations: full saturation, 1/10e, 1/100e.
- Keep the samples at 4 °C for up to 3 days. Crystals of the polyhedrin are very resistant and dense and require long soaks to incorporate the heavy atoms; for other types of crystals the incubation period and concentration of heavy atoms should be determined experimentally. Too short incubation times will not allow the incorporation of proper amounts of the heavy atom, while long incubation times can affect the integrity of the crystals, hence lowering their diffraction quality or even dissolving them. As a starting point, try 3 different incubation times for each heavy atom solution (e.g. 1 min, 1 h and overnight).Successful derivatization is assessed by analysis of diffraction data as briefly described in section 5 and references therein.
- Wash away the excess of heavy atom solution by pelleting the crystals at 4,000 x g for 3 min at room temperature in a microcentrifuge, or the cells at 150 x g for 3 min, removing the supernatant and gently resuspending the crystals or cells in 10 µL of buffer.
- Preparation of micromesh support with purified microcrystals
CAUTION: Use of liquid nitrogen is associated with hazards such as asphyxiation, cold burns, fire in oxygen-enriched atmosphere and explosion. Please consult the risk management and safe work practices of your institution.- Based on the crystal concentration calculated in step 2.1.9, adjust the concentration to 107 crystals/mL by diluting the sample; or by pelleting the crystals at 4,000 x g for 3 min at room temperature in a microcentrifuge, adjusting the final volume by removing supernatant, and gently resuspending the crystals.
- If samples are meant to be frozen in advance, cool down the dry shipper or dewar with liquid nitrogen. Prepare the vials or pucks, as appropriate, and cool them down with liquid nitrogen.
- Resuspend the crystal pellet and apply a 0.5 µL droplet of crystals onto a micromesh by pipetting. Use 700 µm meshes with 25-µm square holes as a starting point. The area of the mesh is not critical but a larger area provides more crystals per mesh and leads to slower evaporation during preparation. The size of holes may need to be optimized to match the size of crystals larger than 25 µm. If required for systematic studies, indexed meshes are available to facilitate a methodical grid scan during data collection.
- Remove most of the excess liquid by blotting with a paper wick. The crystals should be spread evenly on the mesh, without touching each other. Repeat step 3.1.1. for optimization of sample concentration. Proceed quickly and do not remove too much of the liquid. In the absence of cryoprotectant, the liquid evaporates rapidly with the risk of salt crystal formation and/or loss of the film.
- Add cryoprotectant: pipet 0.5 µL of a solution of 50% ethylene glycol in water onto the mesh (the composition of the cryobuffer is to be adapted to the sample; suggestions for optimization of cryoprotectant can be found in reference30). Remove the excess liquid by blotting with a paper wick. By contrast with the previous step, here the remaining film of solvent should be as thin as possible to reduce parallax effects that complicate the process of alignment between the crystal, the beam path and the goniometer; and to minimize background caused by scattering of X-rays by liquid in the beam path.
- Immediately flash-cool the micromesh in liquid nitrogen and transfer to a vial or robot puck, then to a dry shipper or dewar for storage.
- Preparation of micromesh with crystal-containing cells
CAUTION: Use of liquid nitrogen is associated with hazards such as asphyxiation, cold burns, fire in oxygen-enriched atmosphere and explosion. Please consult the risk management and safe work practices of your institution.- Transfer the sorted cells to a 1.5 mL microcentrifuge tube. Based on the cell counts recorded by the cell sorter, adjust the concentration to 107 cells/mL by diluting the sample with PBS; or by pelleting the cells at 150 x g for 3 min at room temperature in a microcentrifuge, adjusting the final volume by removing supernatant, and gently resuspending the cells.
- Mix the cells with an equal volume of trypan blue solution (0.4% w/v) to a final concentration of 0.2 % w/v.
- If samples are meant to be frozen in advance, cool the dry shipper or dewar down with liquid nitrogen. Prepare the vials or pucks, as appropriate, and also cool them down with liquid nitrogen.
- Gently resuspend the cells and pipet 0.5 µL of sorted cells onto a micromesh.
- Remove the excess liquid by blotting with a paper wick. The cells should be spread evenly on the mesh, without touching each other. If not using cryoprotectant (step 3.3.6), the remaining film of solvent should be as thin as possible to reduce parallax effects that complicate the process of alignment between the crystal, the beam path and the goniometer; and to minimize background caused by scattering of X-rays by liquid in the beam path.
- Optionally, add cryoprotectant to the cells: pipet 0.5 µL of 50% ethylene glycol, 50% PBS solution onto the mesh. The composition of the cryobuffer is to be adapted to the sample; suggestions for optimization of cryoprotectant can be found in reference30. Remove the excess liquid by blotting with a paper wick, leaving only a thin film of solvent.
Note: Omission of the cryoprotection step did not alter the quality of X-ray diffraction when crystals of the polyhedrin were analyzed in cellulo, while this step was required for purified crystals20. This only applies to the incubation of the cells with cryoprotectant solution; the sample should still be analyzed at 100K to minimize the effects of radiation damage on the crystals. - Immediately flash-cool the micromesh in liquid nitrogen and transfer to a vial or robot puck, then to a dry shipper or storage dewar, as appropriate.
4. Data Collection
Note: Parameters used for data collection are given as a guide and should be optimized for each crystal type and synchrotron beamline.
- Collect data on a microfocus crystallography beamline (cf. reference 7 for a list of microfocus beamlines and their characteristics). If available, use a collimated beam that matches the size of the crystals.
- For experimental phasing by the Single-wavelength Anomalous Dispersion method (SAD), collect at an energy maximizing the anomalous signal of the heavy atom of choice. For phasing using the Multiple Isomorphous Replacement method, (MIR), collect above the energy of a suitable absorption edge for the heavy atom of choice (available at http://skuld.bmsc.washington.edu/scatter/AS_periodic.html).
- After loading the mesh onto the goniometer, start by centering the micromesh with the beam path using the face-on (convex side) and side-on orientations. Then, with the micromesh face-on, fine-tune the alignment by aligning the center of a particular horizontal lane of the micromesh (for instance, start with the central one). Then collect data along this horizontal lane with only minor readjustments to the alignment. If available, use centering based on rastering at low-dose X-ray diffraction for fine-tuning the alignment (consult the local contact at the synchrotron beamline).
Note: As a limited amount of data can be collected on microcrystals due to radiation damage (typically, 10 to 30 images with 1 ° oscillation), in theory the crystal only needs to be aligned with the beam path for 10-30 °. However, it is critical that the alignment is accurate as the crystal can easily by missed by the microbeam (generally collimated to a diameter of ~10 µm or smaller). - Collect diffraction data until diffraction is lost due to radiation damage. For crystals of the polyhedrin, exposure time of 10 sec is typically used for a 1° oscillation for a flux of approximately 3.6 x 1011 photons/s at 13 keV with a CCD detector (total dose <30 MGy). Optimize these settings depending on the beamline and nature of the crystal used. In particular, low dose and fine slicing are recommended for pixel X-ray detectors.
Note: If using the in cellulo strategy, the cells stained with trypan blue will appear as blue dots against the support background, making them easier to align to the beam. If the collimated microbeam has approximately the same size as the cells (~10 µm), all crystals contained in a cell will be illuminated simultaneously. For cells containing multiple microcrystals, this leads to the observation of multiple diffraction patterns, which can be difficult to process. However, diffraction from one of the crystals often tends to dominate the diffraction pattern and is preferentially indexed during data processing. For beamlines with an X-ray beam significantly smaller that the cell, rastering at very low X-ray flux (>95% attenuation) should be used to center on a single crystal before data collection. - Proceed to the next crystal in lane.
- Once all the crystals of the lane have been tested, proceed to the next lane and repeat the alignment procedure. Proceed until sufficient data has been collected or all crystals have been tested.
- Make sure that the lanes that have been processed are clearly identified so that crystals are not missed or shot twice (if necessary, draw a sketch). Typically, trypan blue turns yellow after the cell has been irradiated, which may be used as a marker.
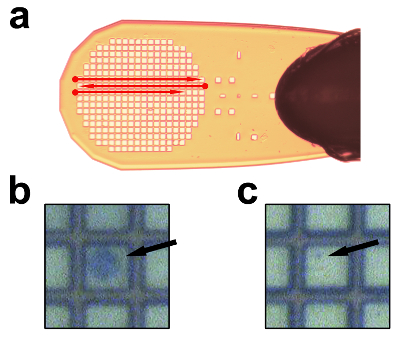
Figure 5: Data Collection Strategy from Samples on Micromesh Support. (a) Strategy of data collection per lane; alignment is done at the beginning of each lane (dots) and data is collected along the lane. (b) Close-up of a trypan blue-stained, crystal-containing cell on a mesh, as seen on the microcrystallography beamline screen. (c) Close-up of a purified polyhedra on a mesh, as seen on the microcrystallography beamline screen. The mesh squares are 25 µm x 25 µm. Please click here to view a larger version of this figure.
5. Data Processing
NOTE: Here, we only mention briefly the steps of data processing that are specific to serial microcrystallography, where data from multiple crystals are merged. Many excellent articles are available describing data processing in depth31,32,33,34,35,36 and structure determination is beyond the scope of this protocol.
- Select the crystal with the best diffraction resolution and quality as a reference.
- Individually process each crystal using standard crystallography packages such as HKL-200031, Mosflm32, or XDS33. To ensure that radiation damage does not deteriorate the quality of the dataset, carefully select the last image with acceptable radiation damage. Cut-off criteria indicating excessive radiation damage are specific of each crystal type, processing practice and the purpose of the experiment. For phasing of the BmCPV1 polyhedrin crystals, data was discarded once the signal-to-noise <I>/<σ(I)> per image dropped below 2 or the Rsym exceeded 50% as reported by HKL-200031.
- For each crystal, define the resolution limit and number of frames and reprocess with the correct parameters to avoid integrating data with little or no signal.
- Scale and merge data from the best crystals to the reference crystal(s) using standard packages such as Scalepack in HKL-200031, SCALA in CCP434,35 or XSCALE in XDS33. Add the data from the different crystals progressively while monitoring the global quality of the dataset, ideally until the dataset reaches an acceptable completeness (>95%). Reject crystals that present a poor isomorphism with the best/reference crystal as detected by differences in unit cell parameters and poor Rmerge/chi-square per image. If too many good quality datasets do not scale with the reference crystal(s), use a program like BLEND36 that carries hierarchical clustering to define the best combination of datasets to merge.
Note: In some space groups, alternate indexing options need to be tested to match the reference crystal. - Proceed to experimental phasing, molecular replacement or refinement using standard crystallography packages.
An overview of both alternative methods for structure determination using in vivo microcrystals is presented (Figure 1). Polyhedra can easily be purified by sonication and centrifugation. Due to their density, they form a layer at the bottom of the tube underneath a layer of debris that can be removed by pipetting (Figure 3a and 3b). The sample is then subjected to several rounds of sonication and washes until a sufficient level of purity is reached as judged from the white and chalky aspect of the pellet (Figure 3c).
For the in cellulo approach, flow cytometry profile of non-infected cells is compared with profile of crystal-containing cells and used to determine which cell population should be selected to sort crystal-containing cells away from the other cells (Figure 4). In this example, cells containing polyhedra have a higher side-scattering than non-infected cells. Other differences in scattering patterns may be observed for other crystal types and gating should be modified accordingly.
Purified microcrystals or crystal-containing cells are pipetted onto a micromesh (Figure 5). Cells stained with trypan blue can be easily visualized using cameras available at most of synchrotron beamlines (Figure 5b), while purified crystals are laborious to identify and align with the X-ray beam due to their small size (Figure 5c). When collecting data, it is best to proceed in a grid pattern in order to minimize the centering procedures, and to avoid exposing twice the same cell or crystal, or missing any (Figure 5a). Data collected should be processed carefully to account for differences in diffraction limits, the effect of radiation damage and a possible lack of isomorphism.
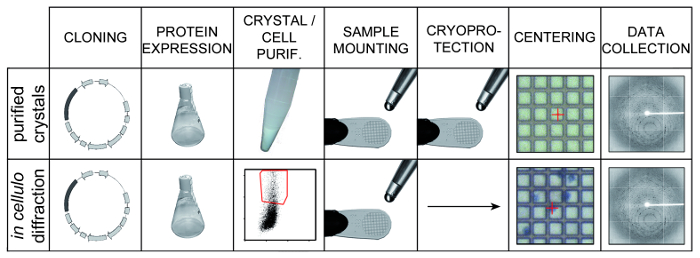
Figure 1: Overview of Two Methods for Structure Determination using In Vivo Microcrystals. Microcrystals are produced in cell culture by over-expression of the protein of interest using a recombinant expression system. In specific cases, microcrystals may also be produced naturally by the cells in particular conditions. The top row shows the classical approach where cells are lysed and crystals are purified by differential centrifugation. Purified microcrystals are pipetted onto a micromesh. After removal of the excess liquid, a cryoprotectant solution is added, excess liquid is blotted again and the mesh is flash-cooled. For X-ray diffraction experiments, crystals are carefully aligned with the X-ray beam, which may be the rate limiting step in data collection. The bottom row presents the in cellulo approach. Cells are sorted by flow-cytometry to specifically select crystal-containing cells. Cells are stained with trypan blue to facilitate visualization and spread onto a micromesh. In this case, the addition of a cryoprotectant is optional. For X-ray diffraction experiments, cells are easily visualized using typical cameras available at synchrotron beamlines facilitating the centering process.
Image of automated liquid chromatography system courtesy of GE Healthcare AB, Uppsala, Sweden. Please click here to view a larger version of this figure.
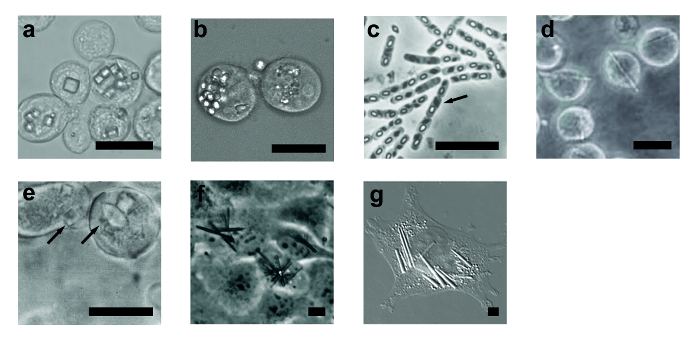
Figure 2: Examples of In Vivo Microcrystals. Selected examples of in vivo crystals: (a) Sf9 cells with cypovirus polyhedra; (b) LD652 cells with entomopoxvirus spheroids; (c) Bacillus thuringiensis with Cry3A crystals (arrow); (d) Sf9 cells with Trypanosoma brucei cathepsin B crystals; (e) Sf21 cells with calcineurin crystals (arrows); (f) COS7 cells monolayer with PKA:Inka1 crystals; (g) CHO cells with IgG crystals. Reproduced from14,18,29,37,39,42, with permission. Scale bar = 10 µm. Please click here to view a larger version of this figure.
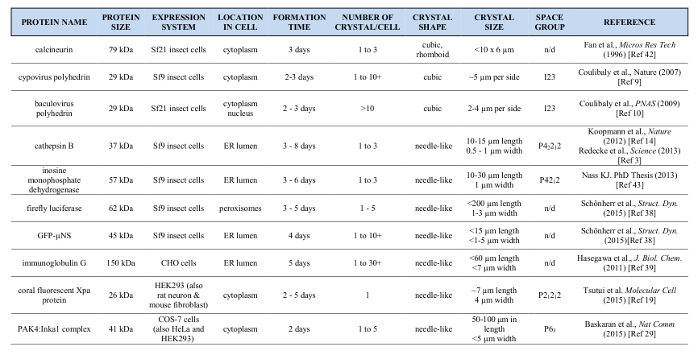
Table 1: Summary of In Vivo Crystals Grown in Cultured Cells Expressing Recombinant Protein. Please click here to view a larger version of this figure.
