The Lambda Select cII Mutation Detection System
Summary
We describe a detailed protocol for the Lambda Select cII mutation assay in cultured cells of transgenic rodents or the corresponding animals treated with a chemical/physical agent of interest. This approach has been widely used for mutagenicity testing of carcinogens in mammalian cells.
Abstract
A number of transgenic animal models and mutation detection systems have been developed for mutagenicity testing of carcinogens in mammalian cells. Of these, transgenic mice and the Lambda (λ) Select cII Mutation Detection System have been employed for mutagenicity experiments by many research groups worldwide. Here, we describe a detailed protocol for the Lambda Select cII mutation assay, which can be applied to cultured cells of transgenic mice/rats or the corresponding animals treated with a chemical/physical agent of interest. The protocol consists of the following steps: (1) isolation of genomic DNA from the cells or organs/tissues of transgenic animals treated in vitro or in vivo, respectively, with a test compound; (2) recovery of the lambda shuttle vector carrying a mutational reporter gene (i.e., cII transgene) from the genomic DNA; (3) packaging of the rescued vectors into infectious bacteriophages; (4) infecting a host bacteria and culturing under selective conditions to allow propagation of the induced cII mutations; and (5) scoring the cII-mutants and DNA sequence analysis to determine the cII mutant frequency and mutation spectrum, respectively.
Introduction
A wide range of transgenic animal models and mutation detection systems have been developed for mutagenicity testing of carcinogens in mammalian cells. Of these, transgenic Big Blue (referred to hereafter as BB) mice and the λ Select cII Mutation Detection System have been employed for mutagenicity experiments by this group and many other research groups worldwide1,2,3,4,5,6,7,8,9. For the past 16 years, we have investigated the mutagenic effects of various chemical and/or physical agents using these transgenic animals or their corresponding embryonic fibroblast cell cultures treated with a test compound, and subsequently analyzed the phenotype and genotype of the cII transgene by the λ Select cII assay and DNA sequencing, respectively10,11,12,13,14,15,16,17,18,19,20,21,22,23,24. The genome of these transgenic animals contains a bacteriophage λ shuttle vector (λLIZ) integrated on chromosome 4 as a multi-copy head-to-tail concatemer1,2,25. The λLIZ shuttle vector carries two mutational reporter genes, namely the lacI and cII transgenes1,2,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47. The λ Select cII assay is based on the recovery of the λLIZ shuttle vectors from the genomic DNA of cells derived from organs/tissues of transgenic animals1,2,25. The recovered λLIZ shuttle vectors are then packaged into λ phage heads capable of infecting an indicator host Escherichia coli. Subsequently, the infected bacteria are grown under selective conditions to allow for scoring and analysis of mutations in the cII transgene1,3.
Here, we describe a detailed protocol for the λ Select cII assay, which consists of isolation of genomic DNA from cells/organs of transgenic animals treated in vitro/in vivo with a test compound, retrieval of the λLIZ shuttle vectors from the genomic DNA, packaging of the vectors into infectious λ phages, infection of the host E. coli with the bacteriophages, identification of the cII-mutants under selective conditions to determine the cII mutant frequency, and DNA sequence analysis to establish the cII mutation spectrum. The protocol can be applied to transgenic mouse/rat cell cultures treated in vitro with a chemical/physical agent of interest, or tissues/organs of the corresponding animals treated in vivo with the test chemical/agent1,2,4,48,49,50,51,52. A schematic presentation of the λ Select cII assay is shown in Figure 1.
Protocol
1. Genomic DNA Isolation from Mouse Embryonic Fibroblasts
NOTE: Primary mouse embryonic fibroblasts are isolated from embryos derived from BB transgenic mice with C57BL/6 genetic background, according to the published protocol53. The starting material for this protocol consists of 1 x 106 to 1 x 107 embryonic fibroblast cells treated with a test compound versus control. The harvesting and counting of these cells using standard methods are described in references10,54,55.
- Prepare buffer A (0.3 M Sucrose, 60 mM KCl, 15 mM NaCl, 60 mM Tris-HCl, pH 8.0, 0.5 mM spermidine, 0.15 mM spermine, and 2 mM EDTA), buffer B (150 mM NaCl and 5 mM EDTA, pH 7.8), and buffer C (20 mM Tris-HCl, pH 8.0, 20 mM NaCl, 20 mM EDTA, and 1% SDS) in advance, and preserve at room temperature (RT) for up to one year54,55.
- Resuspend cells in 2–4 mL buffer A in a 15 mL conical centrifuge tube by repeatedly pipetting up and down using a wide bore 1,000 µL pipette tip.
- Add one volume (2–4 mL) buffer A containing 1% octylphenoxypolyethoxyethanol (e.g., Nonidet P40) using a glass serological pipette.
- Incubate the tube on ice for 5 min.
- Centrifuge the tube at 1,000 x g for 5 min at RT.
- Discard the supernatant and wash the pellet with 10–15 mL buffer A using a glass serological pipette.
- Resuspend the pellet in 2–4 mL buffer B.
- Add one volume (2–4 mL) buffer C containing 600 µg/mL proteinase K.
- Incubate the tube for 3 h at 37 °C.
- Add RNase A to a final concentration of 100 µg/mL.
- Incubate the tube for an additional 1 h at 37 °C.
- Add one volume (2–4 mL) phenol (saturated in 0.1 M Tris-HCl, pH 8.0):chloroform:isoamyl alcohol (25:24:1 vol/vol).
NOTE: Phenol can pose a severe health hazard and must be handled with extreme caution. Phenol is highly corrosive to the skin and readily absorbed through it, and exhibits other effects. When handling phenol, always use double gloving, wear protective eyewear, and work in a chemical fume hood. - Mix well by inverting the tube for 5 min on a tube rotator at 4 °C.
- Centrifuge the tube at 1,000 x g for 5 min at 4 °C.
- Remove the aqueous phase (top layer) to a new tube using a glass serological pipette.
- Repeat phenol:chloroform:isoamyl alcohol extraction 1 to 3 times until the aqueous phase is clear and the interface is no longer turbid.
- Add 1/10 volume (200–400 µL) 3M Sodium acetate, pH 5.2.
- Add 2.5 volume (5–10 mL) 100% ethanol (chilled) and invert the tube gently by hand for 2–3 min.
- Spool the DNA with a glass hook and transfer it to a new tube containing 1–5 mL 70% Ethanol and wash it thoroughly. Alternatively, centrifuge the tube at high speed (3,500 x g) at 4 °C to pellet the DNA and wash it thoroughly with 1–5 mL 70% Ethanol.
- Centrifuge the tube at high speed (3,500 x g) at RT, remove the supernatant, and air-dry the DNA for 10-15 min.
- Dissolve the DNA in 10–100 µL TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.5), and store at 4 °C (for short storage of up to one month) or at -20 °C (for longer storage period). The optimal concentration for DNA for the λ Select cII assay is 0.5–1.0 µg/µL (in TE buffer)56.
2. In Vitro Packaging Reaction
- Per one packaging reaction, add ~5 µg genomic DNA (final volume: 8–12 µL, genomic DNA was isolated in Section 1 from mouse embryonic fibroblasts treated in vitro with a test compound or control) to a microcentrifuge tube containing the first reaction mix (~10 µL).
NOTE: In-house prepared or commercially available λ packaging extracts are used in different laboratories. In the commercial packaging extract kit used here56 (see the Table of Materials), red tubes contained the first reaction mix (~10 µL) and blue tubes contained the second reaction mix (~70 µL for at least 5 reactions). - Incubate the tube for 90 min at 30 °C.
- Add the required volume (~12 µL) of the second reaction mix to the tube.
- Incubate the tube for an additional 90 min at 30 °C.
- Add 1.1 mL of SM buffer to the tube.
NOTE: 1 mL of this solution (i.e., packaging reaction mixture) will be used for screening λ cII-mutants. The remainder will be used for titering.- Prepare SM buffer by mixing 5.8 g NaCl, 2.0 g MgSO4.7H2O, 50 mL 1 M Tris-HCl (pH 7.5), and 5 mL 2% (w/v) gelatin. Add dH2O to a final volume of 1 liter, autoclave for 30 min. Store at room temperature for up to 1 year.
- Vortex the tube containing the packaged DNA sample for 10 s at RT (vigorous vortexing).
- Pulse spin the tube in a microcentrifuge and store on ice until ready to use. If the sample is not going to be used within the same day of packaging, add 50 µL of chloroform per mL of packaged DNA sample, vortex gently, and store at 4 °C for up to 2 weeks.
3. Preparing the E. coli G1250 Bacterial Culture
- At least two days before plating, make a few bacterial streak plates from Escherichia coli (E. coli) G1250 on TB1-kanamycin agar plates.
- Prepare TB1-kanamycin agar plates as follows. Mix 5.0 g NaCl, 10.0 g Casein peptone, and 12.0 g agar. Add 800 mL dH2O. Add 1 mL 0.1% Thiamine hydrochloride. Adjust pH to 7.0 with NaOH or HCl. Add dH2O to a final volume of 1 liter.
- Mix well and autoclave for 30 min. Allow to cool to 55 °C. Add 50.0 mg Kanamycin and mix. Pour into sterile 100-mm petri dishes (20–25 mL per dish). Store plates at 4 °C for up to two weeks.
- Incubate the bacterial streak plates in a stationary 30 °C incubator for at least 24 h.
- One day before plating, combine 10 mL of TB1 liquid medium with 100 µL of 20% (w/v) maltose-1 M MgSO4 solution in a sterile 50 mL screw-cap conical tube.
- Prepare TB1 liquid medium as follows. Mix 5.0 g NaCl and 10.0 g Casein peptone, then add 800 mL dH2O, 1 mL 0.1% Thiamine hydrochloride, and adjust the pH to 7.0 with NaOH or HCl. Then add dH2O to a final volume of 1 liter, mix well, and autoclave for 30 min. Store the medium at RT for up to three months.
- Prepare 20% (w/v) Maltose-1 M MgSO4 as follows. Mix 20.0 g Maltose and 24.6 g MgSO4. 7H2O, then add dH2O to a final volume of 100 mL. Filter sterilize and store at 4 °C for up to 6 months.
- Inoculate the liquid medium with several colonies from the bacterial streak plate using a sterile inoculating loop56.
- Incubate the liquid culture overnight in a 30 °C shaking incubator with vigorous shaking (250–300 rpm).
- On the day of plating, centrifuge the conical tube containing the G1250 liquid culture at 1,500 x g for 10 min at RT to pellet the bacterial cells.
- Discard the supernatant and resuspend the cell pellet in 10 mL of 10 mM MgSO4.
- Measure the absorbance of a 1:10 dilution of the cell suspension at wavelength 600 nm using a UV-Vis spectrophotometer (e.g., 100 µL cell suspension + 900 µL 10 mM MgSO4)56.
- Dilute the cell suspension to a final OD600 of 0.5 with 10 mM MgSO4. The prepared suspension of G1250 E. coli with OD600 = 0.5 is referred to as 'the G1250 plating culture'.
- Store the G1250 plating culture on ice and use within 1-2 h.
4. Plating the Packaged DNA Samples
- Per one packaged DNA sample, prepare sixteen sterile 14 x 100 mm2 round-bottom tubes and sixteen TB1 agar plates. Ten from each set will be used for screening and six for titering (three for Titer 20 and three for Titer 100).
- Prepare TB1 agar plates as follows. Mix 5.0 g NaCl, 10.0 g Casein peptone, and 12.0 g Agar, then add 800 mL dH2O, and 1 mL 0.1% Thiamine hydrochloride. Adjust the pH to 7.0 with NaOH or HCl, and add dH2O to a final volume of 1 L.
- Mix well and autoclave for 30 min. Allow the mixture to cool to 55 °C, then pour it into sterile 100 mm Petri dishes (20–25 mL per dish). Store the plates at 4 °C for up to two weeks.
NOTE: TB1 agar plates should be prepared at least 24 h prior to use.
- Aliquot 200 µL of the G1250 plating culture into each round-bottom tube.
- For titering, make a 1:100 dilution of the packaged DNA sample and mix well by vortexing (i.e., 10 µL packaged DNA sample + 990 µL SM buffer).
- Add 20 µL of the 1:100 dilution to each of the three Titer 20 tubes.
- Add 100 µL of the 1:100 dilution to each of the three Titer 100 tubes.
- For screening, add 100 µL of the 'undiluted' packaged DNA sample to each of the ten screening tubes.
- Process all titering and screening tubes (2 x 3 + 10 = 16 tubes), as follows: mix well by vortexing for approximately 10 s, and then incubate at room temperature for 30 min to allow the host E. coli to adsorb the phages.
- Add 2.5 mL microwaved molten TB1 top agar (cooled to 55 °C) to each titer or screening tube, mix immediately by vortexing (gently), and pouring into the appropriate titer or screening TB1 agar plates.
- Prepare TB1 top agar as follows. Mix 5.0 g NaCl, 10.0 g Casein peptone, and 7.0 g Agar, then add 800 mL dH2O. Add 1 mL 0.1% Thiamine hydrochloride, then adjust the pH to 7.0 with NaOH or HCl. Finally, add dH2O to a final volume of 1 liter.
- Mix well and autoclave for 20–25 min. Store at room temperature for up to three months.
- Prior to use, melt the prepared TB1 top agar in a microwave, mix well, and allow to cool to 55 °C in a water bath.
NOTE: The molten and cooled TB1 top is added to the contents of each titer or screening tube, and after mixing, is poured into the appropriate titer or screening TB1 agar plates.
- Let the plates stand for 15–30 min at room temperature, with the lid ajar to prevent condensation.
- Invert the plates and place the ten screening plates in a stationary 24 °C incubator and incubate for 46–48 h (i.e., selective conditions) and the six titer plates in a stationary 37 °C incubator and incubate for 24 h/overnight (i.e., non-selective conditions).
NOTE: For quality assurance and standardization of results, commercially available control phage solutions containing a mixture of wildtype and mutant cII with known mutant frequency are plated alongside the packaged DNA samples and included in all assay runs56.
5. Examining the Titer and Screening Plates to Determine the cII Mutant Frequency
- Following the 24 h/overnight incubation at 37 °C, count the number of plaques formed in each of the three Titer 20 plates and Titer 100 plates. To more easily identify the plaques, hold the plate next to a white light box and against a dark background with lid removed (see Figure 2).
- Make an average (mean) of the number of plaques counted in each set of Titer 20 plates and Titer 100 plates (triplicate, each).
- Choose the average number from the set of titer plates that falls closest to the range of 50–200 plaques per plate set.
- Calculate the total number of plaques screened in the ten screening plates as follows:
Total plaques screened = (Mean number of plaques in the chosen set of titer plates ÷ Number of µL of dilution used per chosen titer plate) x Dilution factor x Number of µL of packaged DNA sample per screening plate [100 µL/plate] x Number of screening plates [10].
NOTE: For example, if the mean number of plaques per plate in the set of Titer 20 plates is 128 and the corresponding number in the set of Titer 100 plates is 478, the number 128 will be used for calculation of the total number of plaques screened, as follows:
(128 ÷ 20) x 100 [dilution factor] x 100 [µL/plate] x 10 [plates] = 640,000
This is equivalent to multiplying the mean number of plaques by 5,000 or 1,000, if the average number is based on counts from Titer 20 plates or Titer 100 plates, respectively. - Following 46–48 h incubation at 24 °C, count the total number of plaques formed in the ten screening plates (follow the instructions provided in Section 5.1).
NOTE: The cII mutant frequency is calculated by dividing the total number of plaques formed in the screening plates by the total number of plaques screened. Using the above example, if the total number of plaques counted in the ten screening plates is 112, the cII mutant frequency for this DNA sample would be 112 ÷ 640,000 = 17.5 x 10-5.
6. Verification of the Putative λ cII Mutants, PCR Amplification, and DNA Sequencing
- Core the plaque in question with a sterile, wide-bore pipet tip and expel it (by pipetting up and down) into a sterile microcentrifuge tube containing 500 µL of sterile SM buffer.
- Incubate for at least 2 h at room temperature, or at 4 °C overnight, to allow the phage particles to elute from the agar plug.
- In a sterile 14 x 100 mm2 round-bottom tube, mix 200 µL of the G1250 plating culture with 1 µL of the cored phage solution, and incubate for 30 min at RT.
- Plate the sample using 2.5 mL of 55 °C molten TB1 top agar and incubate the plate at 24 °C for 46–48 h (selective conditions), as described in Sections 4.8–4.10.
- Once the secondary plaques have formed, use a pipette tip to carefully pick a single well-isolated verified λ cII-mutant plaque (avoid touching the lower agar).
NOTE: Replating the putative cII mutant plaques serves two purposes: (i) plating artefacts may sometimes be mistaken for small-size plaques; and (ii) an agarose core taken from a screening plate may contain non-mutant phage(s) together with a mutant phage. Secondary plaques from a low-density replating will provide an uncontaminated mutant template for PCR and subsequent DNA sequence analysis. - Transfer the plaque to a microcentrifuge tube containing 25 µL double-distilled water by pipetting up and down.
- Place the tube in boiling water for 5 min.
- Centrifuge at maximum speed (18,000 x g) for 3 minutes at RT.
- Transfer 10 µL of the supernatant immediately to a new microcentrifuge tube containing 40 µL of a PCR mastermix in which the final concentrations of the reagents are 1 x Taq PCR buffer, 10 pmol each of the forward and reverse primers, 12.5 nmol of each dNTP, and 2.5 U of Taq DNA polymerase.
NOTE: The forward and reverse primers are as follows: 5'-CCACACCTATGGTGTATG-3' (positions -68 to -50 relative to the cII start codon) and 5'-CCTCTGCCGAAGTTGAGTAT-3' (positions +345 to +365 relative to the cII start codon), respectively. - Amplify the template using the following cycling parameters: a 3 min denaturation at 95 °C, followed by 30 cycles of 30 s at 95 °C, 1 min at 60 °C, and 1 min at 72 °C, with a final extension of 10 min at 72 °C.
- Purify the 432 bp PCR product containing the cII gene and flanking regions using commercially available PCR purification kits, according to the manufacturer's instructions.
- Perform DNA sequencing using an appropriate sequencing platform, and analyze the resulting DNA sequences to detect mutations in the cII transgene (see the note below).
NOTE: This is best achieved by alignment with the reference cII sequence using software, such as the web-based T-Coffee sequence alignment server. For instructions on how to use the program, visit: http://tcoffee.crg.cat/
Representative Results
Depending on data distribution, parametric or non-parametric tests are used to determine the significance of difference in the cII mutant frequency between treatment and control groups (i.e., induced versus spontaneous mutant frequencies). Comparison of the induced cII mutant frequencies across different treatment groups is made by various (pairwise) statistical tests, as applicable. The hypergeometric test of Adams and Skopek is commonly used to compare the overall induced- and spontaneous mutation spectra57, although other tests, such as the χ2 test or Analysis of Variance (ANOVA), can also be used to compare the frequency of each specific type of mutation (e.g., transition, transversion, insertion, or deletion) between the induced- and control mutation spectra, or among various mutation spectra induced by different chemicals/agents or varying doses of the same chemical/agent.
Figure 3 is a compilation of mutant frequency data from published studies in which we have demonstrated that the extent of increase in relative cII mutant frequency in mouse embryonic fibroblasts treated with various chemicals and/or physicalagents may vary from a few- to several hundred-fold, depending on the mutagenic 'potency' of the test compound. Statistically significant fold-increases in the cII mutant frequency are shown for mouse embryonic fibroblasts treated with acrylamide12, glycidamide14, aflatoxinB1(AFB1)22, tamoxifen18, δ-aminolevulinic acid (δ-ALA) plus low dose ultraviolet light A (UVA: λ >320−400 nm)15, benzo(a)pyrene diol epoxide(B(a)PDE)19, and equilethal doses of UVA, UVB (λ =2 80−320 nm), and simulated sunlight UV (SSL) 21 (see Figure 3).
Figure 4 is a demonstration of the 'sequence-specificity' of mutations in which we have shown the induction of specific types of mutation in the cII transgene in mouseembryonicfibroblasts irradiated with UVBrelativetocontrol23.The UVB-induced mutation spectrum is characterized by significant increases in relative frequency of single- or tandem C→T transitions at pyrimidine dinucleotides.
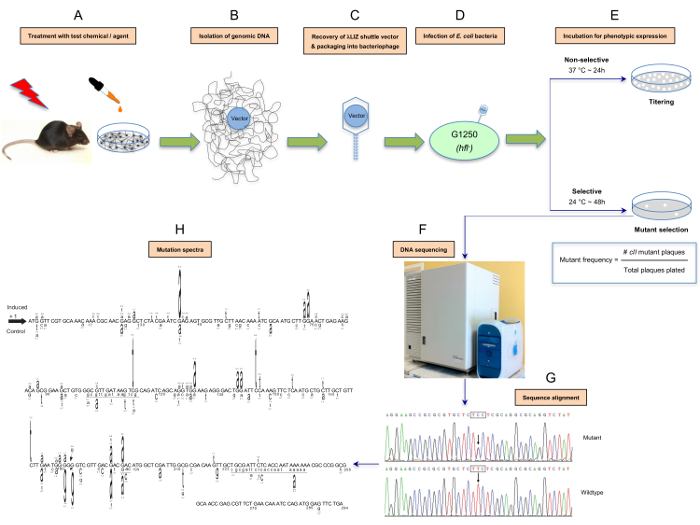
Figure 1: Schematic presentation of the λ Select cII assay. The assay is based on the retrieval of the λLIZ shuttle vectors, which contain the cII transgene as a mutational reporter gene,from the genomic DNA of cultured cells derived from transgenic rodents treated in vitro with a test compound or tissues/organs of the corresponding animals treated in vivo with the tested chemical/agent (A and B). The rescued vectors are packaged into λ phage heads that can infect an appropriate host E. coli (C and D). The infected bacteria are then grown under selective conditions to allow for scoring and analysis of mutations in the cII transgene1,2,3,25,52 (E). Determination of the induced cII mutant frequency and establishment of the mutation spectrum by DNA sequencing are outlined in F and G. The induced- and spontaneous mutation spectra are visualized in different formats. For illustration purposes, we have highlighted a format in which the induced cII mutations are typed above the reference sequence, whereas the spontaneous mutations (control) are typed below the reference sequence (H). The height of a mutated base represents its frequency of mutations (i.e., the higher the base, the more frequently mutated). Numbers above a mutated base indicate the percentage frequency of mutations in that base. Deleted bases are underlined. Inserted bases are shown with an arrow. Numbers below the bases are reference nucleotide positions. Data are from a published study23. Please click here to view a larger version of this figure.
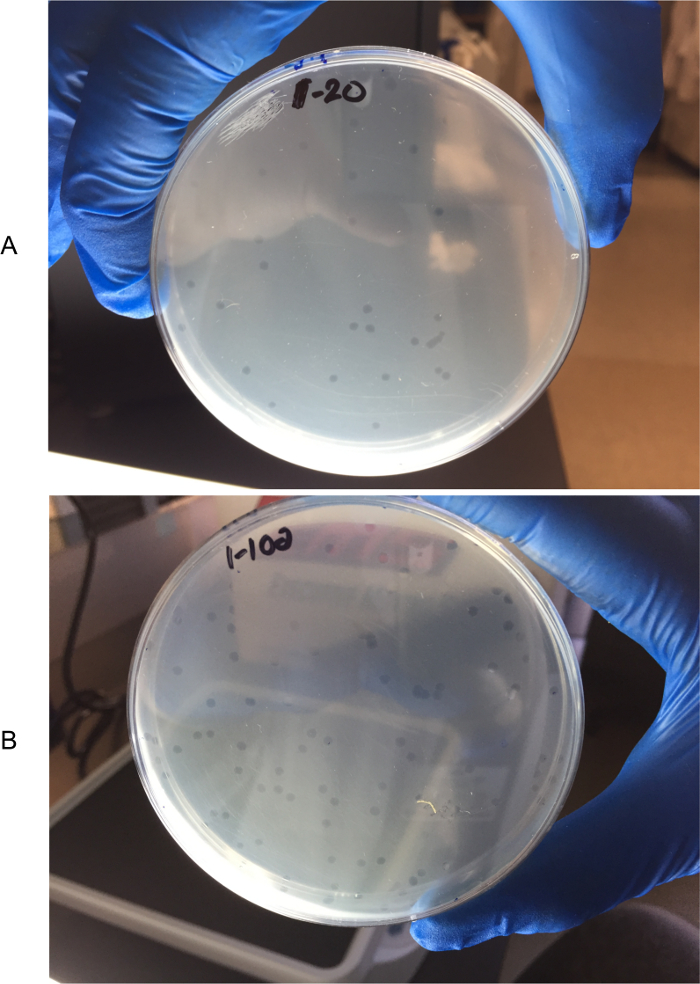
Figure 2: Counting plaques in titer plates. To more easily identify the plaques, plates are held next to a white light box and against a dark background with lids removed. Titer 20 plate (A) and Titer 100 plate (B). Please click here to view a larger version of this figure.
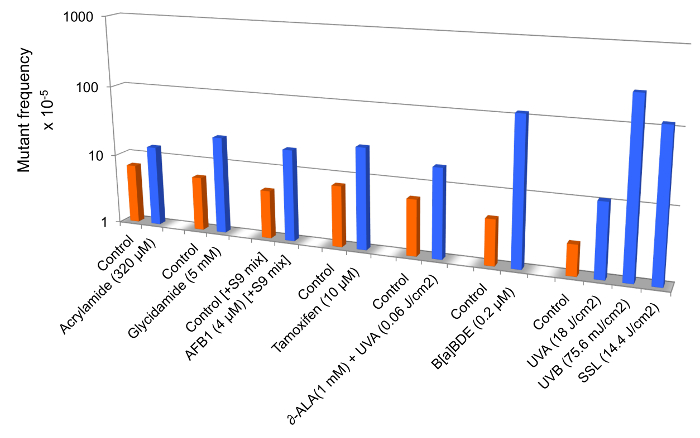
Figure 3: Mutant frequencies of the cII transgene in mouse embryonic fibroblasts treated with various chemicals and/or physical agents in comparison to controls. Data are from published studies on acrylamide12, glycidamide14, aflatoxinB1(AFB1)22, tamoxifen18, δ-aminolevulinic acid (δ-ALA) plus low dose ultraviolet light A (UVA: λ >320−400 nm)15, benzo(a)pyrenediol epoxide (B(a)PDE)19, and equilethal doses of UVA, UVB (λ = 280−320 nm), and simulated sunlight UV (SSL)21.To efficiently metabolize tamoxifen in mouse embryonic fibroblast cells, we used the S9-activation system (S9 mix) consisting of Aroclor 1,254-induced rat liver preparations and cofactor reagents22. All differences between treated- and control samples are statistically significant at p <0.05. Please click here to view a larger version of this figure.
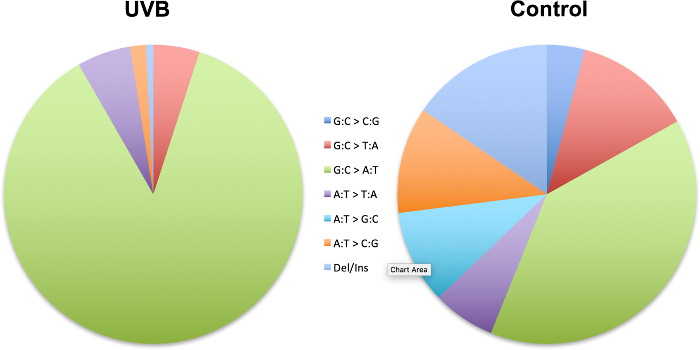
Figure 4: Mutation spectra of the cII transgene in mouse embryonic fibroblasts irradiated with UVB relative to control. Data are from a published study23.The strand mirror counterparts of all transitions (e.g., G→A and C→T) and transversions (e.g., G→T and C→A or G→C and C→G) are combined. Ins: insertion; Del: deletion. The UVB-induced mutation spectrum is characterized by significant increases in relative frequency of single- or tandem C→T transitions at pyrimidine dinucleotides. Please click here to view a larger version of this figure.
Discussion
The λ Select cII assay is used for detection of mutations in the cII transgene recovered from the genomic DNA of cells derived from organs/tissues of BB rodents3. The genome of these transgenic animals contains multiple tandem copies of the chromosomally integrated λLIZ shuttle vector, which carries the cII (294 bp) and lacI (1,080 bp) transgenes, as the mutational reporter genes1,2,25. The λ Select cII assay is based on the retrieval of the λLIZ shuttle vectors from the genomic DNA of cells/tissues of the transgenic animals, followed by packaging of the rescued vectors into λ phage heads that can infect an appropriate host E. coli. Subsequently, the infected bacteria are grown under selective conditions to allow for scoring and analysis of mutations in the cII transgene (see Figure 1)1,3. The λ Select cII assay has been used extensively for mutagenicity testing of a wide range of chemicals and/or physical agents (reviewed in references2,49). The assay has been successfully applied to transgenic mouse/rat cell cultures treated in vitro with various chemicals and/or physical agents, and the tissues/organs of the corresponding animals treated in vivo with different test chemicals/agents4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,34,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75.
The λ Select cII assay in cultured cells of transgenic rodents treated with a test compound represents, in many ways, a viable alternative to in vivo mutagenicity experiments in the corresponding animals treated with the tested chemical/agent3. As a general rule, the in vitro models offer significant advantages over their counterpart in vivo animal models, as they are much less labor intensive and costly, require far less time to be completed, and most importantly, do not involve the direct use of the animals2,50,52. At the same time, the in vitro models may not fully recapitulate all aspects of mutagenesis due to differences in the pharmacokinetic and pharmacodynamic properties of chemicals between the cultured cells in vitro and experimental animals in vivo2,3. For example, chemicals whose route of exposure is inhalation (e.g., cigarette smoke or electronic cigarette vapor) can only be made amenable to in vitro testing in cell cultures after they are converted from gaseous or vapor forms to liquid or condensate, which complicates their pharmacokinetics. Also, an incomplete or absent metabolic capacity of cultured cells in vitro to convert certain chemicals into DNA-reactive species may not represent DNA-damage driven mutagenicity in animals exposed in vivo to genotoxic chemicals2,3. Although, this drawback may be compensated for, to varying extents, by the addition of an external metabolic activation system (i.e., S9 mix) to the in vitro cell culture models22.
Furthermore, the replication of real life human exposure to genotoxic chemicals/agents is more limited with in vitro cell culture models than with experimental animals in vivo3. Generally, humans are exposed to chronic doses of genotoxic agents over a span of several years to a few decades76,77,78. The finite lifespan of cells in culture, as compared to the relatively longer lifetime of rodents (i.e., days/weeks versus a few years) makes modeling of human exposure to genotoxins more challenging in the former models2,3. Nonetheless, mutagenicity analysis with in vitro cell culture models can provide an initial indication of the genotoxic potential of a given chemical/agent(s), and the results can be used as a guide to design 'refined' in vivo experiments which feature a 'reduced' number of animals2,3.
In conclusion, the λ Select cII assay in cultured cells of transgenic rodents treated with a test compound, or the corresponding animals treated with the tested chemical/agent, is a valuable approach for mutagenicity testing. We have successfully used the approach, as have other research groups throughout the world4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,34,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75. More recently, we have expanded the applications of this approach by developing a new technique in which a modification of the λ Select cII assay together with next-generation sequencing enables high throughput analysis of mutations in a time-, cost-, and labor-effective manner23.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We would like to acknowledge the contributions of all colleagues and collaborators to our original studies, whose results have been referred to in this manuscript (for illustrative purposes). The authors' work is supported by grants from the National Institute of Dental and Craniofacial Research of the National Institutes of Health (1R01DE026043) to AB and from the University of California Tobacco-Related Disease Research Program to AB (TRDRP-26IR-0015) and ST (TRDRP-25IP-0001). The sponsors of the study had no role in study design, data collection, data analysis, data interpretation, writing of the report, or in the decision to submit for publication.
Materials
| Agar | MO Bio Laboratories, Inc. | 12112-05 | Bacteriological grade |
| BigDye Terminator v3.1 Cycle Sequencing Kit | Thermo Fisher Scientific | 4337455 | None |
| Casein Peptone | Alfa Aesar | H26557 | None |
| Gelatine | J. T. Baker | 2124-01 | Powder |
| Glycerol | Fisher Scientific | BP 229-1 / M-13750 | None |
| LB Agar | Fisher Scientific | BP 9724-500 | None |
| QIAquick PCR purification kit | Qiagen | 8104 | 50 PCR purification reactions |
| Sodium Acetate Trihydrate | Fisher Scientific | M-15756 | None |
| Taq5000 DNA Polymerase | Qiagen | 201207 | None |
| Thiamine Hydrochloride | Macron Fine Chemicals | 2722-57 | None |
| Transpack Packaging Extract | Stratagene Corp., Acquired by BioReliance | Sigma-Aldrich Corp. | 200223 | 50 packaging reactions |
| Tris Base | Fisher Scientific | BP 152-1 / EC 201-064-4 | None |
| Trypton | Biosciences | RC-110 | None |
References
- Jakubczak, J. L., et al. Analysis of genetic instability during mammary tumor progression using a novel selection-based assay for in vivo mutations in a bacteriophage lambda transgene target. Proc Natl Acad Sci U S A. 93 (17), 9073-9078 (1996).
- Lambert, I. B., Singer, T. M., Boucher, S. E., Douglas, G. R. Detailed review of transgenic rodent mutation assays. Mutat Res. 590 (1-3), 1-280 (2005).
- Besaratinia, A., Pfeifer, G. P. Investigating human cancer etiology by DNA lesion footprinting and mutagenicity analysis. Carcinogenesis. 27 (8), 1526-1537 (2006).
- Watson, D. E., Cunningham, M. L., Tindall, K. R. Spontaneous and ENU-induced mutation spectra at the cII locus in Big Blue Rat2 embryonic fibroblasts. Mutagenesis. 13 (5), 487-497 (1998).
- Erexson, G. L., Watson, D. E., Tindall, K. R. Characterization of new transgenic Big Blue(R) mouse and rat primary fibroblast cell strains for use in molecular toxicology studies. Environ Mol Mutagen. 34 (2-3), 90-96 (1999).
- Erexson, G. L., Tindall, K. R. Micronuclei and gene mutations in transgenic big Blue((R)) mouse and rat fibroblasts after exposure to the epoxide metabolites of 1, 3-butadiene. Mutat Res. 472 (1-2), 105-117 (2000).
- McDiarmid, H. M., Douglas, G. R., Coomber, B. L., Josephy, P. D. 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced mutagenesis in cultured Big Blue rat mammary epithelial and fibroblast cells. Environ Mol Mutagen. 39 (2-3), 245-253 (2002).
- Papp-Szabo, E., Douglas, G. R., Coomber, B. L., Josephy, P. D. Mutagenicity of the oral carcinogen 4-nitroquinoline-1-oxide in cultured BigBlue rat tongue epithelial cells and fibroblasts. Mutat Res. 522 (1-2), 107-117 (2003).
- Guichard, Y., et al. In Vitro Study of Mutagenesis Induced by Crocidolite-Exposed Alveolar Macrophages NR8383 in Cocultured Big Blue Rat2 Embryonic Fibroblasts. J Toxicol. , 323828 (2010).
- Besaratinia, A., Bates, S. E., Pfeifer, G. P. Mutational signature of the proximate bladder carcinogen N-hydroxy-4-acetylaminobiphenyl: inconsistency with the p53 mutational spectrum in bladder cancer. Cancer Res. 62 (15), 4331-4338 (2002).
- Besaratinia, A., Pfeifer, G. P. Enhancement of the mutagenicity of benzo(a)pyrene diol epoxide by a nonmutagenic dose of ultraviolet A radiation. Cancer Res. 63 (24), 8708-8716 (2003).
- Besaratinia, A., Pfeifer, G. P. Weak yet distinct mutagenicity of acrylamide in mammalian cells. J Natl Cancer Inst. 95 (12), 889-896 (2003).
- Besaratinia, A., Pfeifer, G. P. Biological consequences of 8-methoxypsoralen-photoinduced lesions: sequence-specificity of mutations and preponderance of T to C and T to a mutations. J Invest Dermatol. 123 (6), 1140-1146 (2004).
- Besaratinia, A., Pfeifer, G. P. Genotoxicity of acrylamide and glycidamide. J Natl Cancer Inst. 96 (13), 1023-1029 (2004).
- Besaratinia, A., Bates, S. E., Synold, T. W., Pfeifer, G. P. Similar mutagenicity of photoactivated porphyrins and ultraviolet A radiation in mouse embryonic fibroblasts: involvement of oxidative DNA lesions in mutagenesis. Biochemistry. 43 (49), 15557-15566 (2004).
- Yoon, J. H., et al. DNA damage, repair, and mutation induction by (+)-Syn and (-)-anti-dibenzo[a,l]pyrene-11,12-diol-13,14-epoxides in mouse cells. Cancer Res. 64 (20), 7321-7328 (2004).
- Besaratinia, A., Synold, T. W., Xi, B., Pfeifer, G. P. G-to-T transversions and small tandem base deletions are the hallmark of mutations induced by ultraviolet a radiation in mammalian cells. Biochemistry. 43 (25), 8169-8177 (2004).
- Besaratinia, A., Pfeifer, G. P. Investigating DNA adduct-targeted mutagenicity of tamoxifen: preferential formation of tamoxifen-DNA adducts in the human p53 gene in SV40 immortalized hepatocytes but not endometrial carcinoma cells. Biochemistry. 44 (23), 8418-8427 (2005).
- Kim, S. I., Pfeifer, G. P., Besaratinia, A. Lack of mutagenicity of acrolein-induced DNA adducts in mouse and human cells. Cancer Res. 67 (24), 11640-11647 (2007).
- Besaratinia, A., Kim, S. I., Bates, S. E., Pfeifer, G. P. Riboflavin activated by ultraviolet A1 irradiation induces oxidative DNA damage-mediated mutations inhibited by vitamin C. Proc Natl Acad Sci U S A. 104 (14), 5953-5958 (2007).
- Besaratinia, A., Kim, S. I., Pfeifer, G. P. Rapid repair of UVA-induced oxidized purines and persistence of UVB-induced dipyrimidine lesions determine the mutagenicity of sunlight in mouse cells. FASEB J. 22 (7), 2379-2392 (2008).
- Besaratinia, A., Kim, S. I., Hainaut, P., Pfeifer, G. P. In vitro recapitulating of TP53 mutagenesis in hepatocellular carcinoma associated with dietary aflatoxin B1 exposure. Gastroenterology. 137 (3), (2009).
- Besaratinia, A., et al. A high-throughput next-generation sequencing-based method for detecting the mutational fingerprint of carcinogens. Nucleic Acids Res. 40 (15), e116 (2012).
- Tommasi, S., Bates, S. E., Behar, R. Z., Talbot, P., Besaratinia, A. Limited mutagenicity of electronic cigarettes in mouse or human cells in vitro. Lung Cancer. 112, 41-46 (2017).
- Dycaico, M. J., et al. The use of shuttle vectors for mutation analysis in transgenic mice and rats. Mutat Res. 307 (2), 461-478 (1994).
- Davies, R., et al. Mutational spectra of tamoxifen-induced mutations in the livers of lacI transgenic rats. Environ Mol Mutagen. 28 (4), 430-433 (1996).
- de Boer, J. G., et al. Spectrum of spontaneous mutations in liver tissue of lacI transgenic mice. Environ Mol Mutagen. 30 (3), 273-286 (1997).
- de Boer, J. G., Mirsalis, J. C., Provost, G. S., Tindall, K. R., Glickman, B. W. Spectrum of mutations in kidney, stomach, and liver from lacI transgenic mice recovered after treatment with tris(2,3-dibromopropyl)phosphate. Environ Mol Mutagen. 28 (4), 418-423 (1996).
- Dycaico, M. J., et al. Species-specific differences in hepatic mutant frequency and mutational spectrum among lambda/lacI transgenic rats and mice following exposure to aflatoxin B1. Carcinogenesis. 17 (11), 2347-2356 (1996).
- Skopek, T. R., Kort, K. L., Marino, D. R. Relative sensitivity of the endogenous hprt gene and lacI transgene in ENU-treated Big Blue B6C3F1 mice. Environ Mol Mutagen. 26 (1), 9-15 (1995).
- Skopek, T. R., et al. Mutagenic response of the endogenous hprt gene and lacI transgene in benzo[a]pyrene-treated Big Blue B6C3F1 mice. Environ Mol Mutagen. 28 (4), 376-384 (1996).
- Erfle, H. L., et al. An efficient laboratory protocol for the sequencing of large numbers of lacI mutants recovered from Big Blue transgenic animals. Environ Mol Mutagen. 28 (4), 393-396 (1996).
- Gu, M., Ahmed, A., Wei, C., Gorelick, N., Glickman, B. W. Development of a lambda-based complementation assay for the preliminary localization of lacI mutants from the Big Blue mouse: implications for a DNA-sequencing strategy. Mutat Res. 307 (2), 533-540 (1994).
- Harbach, P. R., Zimmer, D. M., Filipunas, A. L., Mattes, W. B., Aaron, C. S. Spontaneous mutation spectrum at the lambda cII locus in liver, lung, and spleen tissue of Big Blue transgenic mice. Environ Mol Mutagen. 33 (2), 132-143 (1999).
- Kohler, S. W., et al. Spectra of spontaneous and mutagen-induced mutations in the lacI gene in transgenic mice. Proc Natl Acad Sci U S A. 88 (18), 7958-7962 (1991).
- Mittelstaedt, R. A., et al. Comparison of the types of mutations induced by 7,12-dimethylbenz[a]anthracene in the lacI and hprt genes of Big Blue rats. Environ Mol Mutagen. 31 (2), 149-156 (1998).
- Monroe, J. J., Kort, K. L., Miller, J. E., Marino, D. R., Skopek, T. R. A comparative study of in vivo mutation assays: analysis of hprt, lacI, cII/cI and as mutational targets for N-nitroso-N-methylurea and benzo[a]pyrene in Big Blue mice. Mutat Res. 421 (1), 121-136 (1998).
- Morrison, V., Ashby, J. A preliminary evaluation of the performance of the Muta Mouse (lacZ) and Big Blue (lacI) transgenic mouse mutation assays. Mutagenesis. 9 (4), 367-375 (1994).
- Okonogi, H., et al. Agreement of mutational characteristics of heterocyclic amines in lacI of the Big Blue mouse with those in tumor related genes in rodents. Carcinogenesis. 18 (4), 745-748 (1997).
- Provost, G. S., Mirsalis, J. C., Rogers, B. J., Short, J. M. Mutagenic response to benzene and tris(2,3-dibromopropyl)-phosphate in the lambda lacI transgenic mouse mutation assay: a standardized approach to in vivo mutation analysis. Environ Mol Mutagen. 28 (4), 342-347 (1996).
- Shane, B. S., et al. LacI mutation spectra following benzo[a]pyrene treatment of Big Blue mice. Carcinogenesis. 21 (4), 715-725 (2000).
- Shane, B. S., Lockhart, A. M., Winston, G. W., Tindall, K. R. Mutant frequency of lacI in transgenic mice following benzo[a]pyrene treatment and partial hepatectomy. Mutat Res. 377 (1), 1-11 (1997).
- Shephard, S. E., Sengstag, C., Lutz, W. K., Schlatter, C. Mutations in liver DNA of lacI transgenic mice (Big Blue) following subchronic exposure to 2-acetylaminofluorene. Mutat Res. 302 (2), 91-96 (1993).
- Stiegler, G. L., Stillwell, L. C. Big Blue transgenic mouse lacI mutation analysis. Environ Mol Mutagen. 22 (3), 127-129 (1993).
- Walker, V. E., et al. Frequency and spectrum of ethylnitrosourea-induced mutation at the hprt and lacI loci in splenic lymphocytes of exposed lacI transgenic mice. Cancer Res. 56 (20), 4654-4661 (1996).
- Young, R. R., Rogers, B. J., Provost, G. S., Short, J. M., Putman, D. L. Interlaboratory comparison: liver spontaneous mutant frequency from lambda/lacI transgenic mice (Big Blue) (II). Mutat Res. 327 (1-2), 67-73 (1995).
- Zimmer, D. M., Zhang, X. B., Harbach, P. R., Mayo, J. K., Aaron, C. S. Spontaneous and ethylnitrosourea-induced mutation fixation and molecular spectra at the lacI transgene in the Big Blue rat-2 embryo cell line. Environ Mol Mutagen. 28 (4), 325-333 (1996).
- Swiger, R. R., et al. The cII locus in the MutaMouse system. Environ Mol Mutagen. 34 (2-3), 201-207 (1999).
- Heddle, J. A., Martus, H. J., Douglas, G. R. Treatment and sampling protocols for transgenic mutation assays. Environ Mol Mutagen. 41 (1), 1-6 (2003).
- Thybaud, V., et al. In vivo transgenic mutation assays. Mutat Res. 540 (2), 141-151 (2003).
- Manjanatha, M. G., Cao, X., Shelton, S. D., Mittelstaedt, R. A., Heflich, R. H. In vivo cII, gpt, and Spi(-) gene mutation assays in transgenic mice and rats. Methods Mol Biol. 1044, 97-119 (2013).
- Swiger, R. R. Quantifying in vivo somatic mutations using transgenic mouse model systems. Methods Mol Biol. 1105, 271-282 (2014).
- Tommasi, S., Besaratinia, A., Wilczynski, S. P., Pfeifer, G. P. Loss of Rassf1a enhances p53-mediated tumor predisposition and accelerates progression to aneuploidy. Oncogene. 30 (6), 690-700 (2011).
- Saluz, H. P., Jost, J. P. . A Laboratory Guide to Genomic Sequencing. , (1987).
- Wijnholds, J., Philipsen, J. N., Ab, G. Tissue-specific and steroid-dependent interaction of transcription factors with the oestrogen-inducible apoVLDL II promoter in vivo. EMBO J. 7 (9), 2757-2763 (1988).
- . λ Select-cII Mutation Detection System for Big Blue Rodents. INSTRUCTION MANUAL; Catalog #720120; Revision B.0 Available from: https://www.agilent.com/cs/library/usermanuals/public/720120.pdf (2018)
- Adams, W. T., Skopek, T. R. Statistical test for the comparison of samples from mutational spectra. J Mol Biol. 194 (3), 391-396 (1987).
- Kim, S. I., Yoon, J. I., Tommasi, S., Besaratinia, A. New experimental data linking secondhand smoke exposure to lung cancer in nonsmokers. FASEB J. 26 (5), 1845-1854 (2012).
- Yoon, J. I., Kim, S. I., Tommasi, S., Besaratinia, A. Organ specificity of the bladder carcinogen 4-aminobiphenyl in inducing DNA damage and mutation in mice. Cancer Prev Res (Phila). 5 (2), 299-308 (2012).
- Boyiri, T., et al. Mammary carcinogenesis and molecular analysis of in vivo cII gene mutations in the mammary tissue of female transgenic rats treated with the environmental pollutant 6-nitrochrysene. Carcinogenesis. 25 (4), 637-643 (2004).
- Chen, T., et al. Mutations induced by alpha-hydroxytamoxifen in the lacI and cII genes of Big Blue transgenic rats. Carcinogenesis. 23 (10), 1751-1757 (2002).
- Chen, T., et al. 4-Aminobiphenyl induces liver DNA adducts in both neonatal and adult mice but induces liver mutations only in neonatal mice. Int J Cancer. 117 (2), 182-187 (2005).
- Crabbe, R. A., Hill, K. A. Heart tissue of harlequin (hq)/Big Blue mice has elevated reactive oxygen species without significant impact on the frequency and nature of point mutations in nuclear DNA. Mutat Res. 691 (1-2), 64-71 (2010).
- Hernandez, L. G., Heddle, J. A. A carcinogenic western diet does not induce somatic mutations in various target tissues of transgenic C56BL/6 mice. Mutat Res. 570 (2), 185-196 (2005).
- Manjanatha, M. G., et al. Dose and temporal evaluation of ethylene oxide-induced mutagenicity in the lungs of male big blue mice following inhalation exposure to carcinogenic concentrations. Environ Mol Mutagen. 58 (3), 122-134 (2017).
- Manjanatha, M. G., et al. Evaluation of mutagenic mode of action in Big Blue mice fed methylphenidate for 24 weeks. Mutat Res. 680 (1-2), 43-48 (2009).
- McDaniel, L. P., et al. Mutagenicity and DNA adduct formation by aristolochic acid in the spleen of Big Blue(R) rats. Environ Mol Mutagen. 53 (5), 358-368 (2012).
- Mei, N., Heflich, R. H., Moore, M. M., Chen, T. Age-dependent sensitivity of Big Blue transgenic mice to the mutagenicity of N-ethyl-N-nitrosourea (ENU) in liver. Mutat Res. 572 (1-2), 14-26 (2005).
- Mei, N., et al. The genotoxicity of acrylamide and glycidamide in big blue rats. Toxicol Sci. 115 (2), 412-421 (2010).
- Nay, S. L., Lee, D. H., Bates, S. E., O’Connor, T. R. Alkbh2 protects against lethality and mutation in primary mouse embryonic fibroblasts. DNA Repair (Amst). 11 (5), 502-510 (2012).
- Singh, V. K., Ganesh, L., Cunningham, M. L., Shane, B. S. Comparison of the mutant frequencies and mutation spectra of three non-genotoxic carcinogens, oxazepam, phenobarbital, and Wyeth 14,643, at the lambdacII locus in Big Blue transgenic mice. Biochem Pharmacol. 62 (6), 685-692 (2001).
- Stuart, G. R., et al. Interpretation of mutational spectra from different genes: analyses of PhIP-induced mutational specificity in the lacI and cII transgenes from colon of Big Blue rats. Mutat Res. 452 (1), 101-121 (2000).
- Terrell, A. N., et al. Mutagenicity of furan in female Big Blue B6C3F1 mice. Mutat Res Genet Toxicol Environ Mutagen. 770, 46-54 (2014).
- Thompson, C. M., et al. Assessment of the mutagenic potential of Cr(VI) in the oral mucosa of Big Blue(R) transgenic F344 rats. Environ Mol Mutagen. 56 (7), 621-628 (2015).
- Wang, J., Liu, X., Heflich, R. H., Chen, T. Time course of cII gene mutant manifestation in the liver, spleen, and bone marrow of N-ethyl-N-nitrosourea-treated Big Blue transgenic mice. Toxicol Sci. 82 (1), 124-128 (2004).
- LeBlanc, G. A., Bain, L. J. Chronic toxicity of environmental contaminants: sentinels and biomarkers. Environ Health Perspect. 105, 65-80 (1997).
- Arlt, V. M., et al. Aristolochic acid mutagenesis: molecular clues to the aetiology of Balkan endemic nephropathy-associated urothelial cancer. Carcinogenesis. 28 (11), 2253-2261 (2007).
- Besaratinia, A., Pfeifer, G. P. Second-hand smoke and human lung cancer. Lancet Oncol. 9 (7), 657-666 (2008).
