Benchtop Immobilized Metal Affinity Chromatography, Reconstitution and Assay of a Polyhistidine Tagged Metalloenzyme for the Undergraduate Laboratory
Summary
Here we present a protocol for the benchtop immobilized metal affinity chromatography purification and subsequent reconstitution of a polyhistidine tagged, non-heme iron binding dioxygenase suitable for the undergraduate teaching laboratory.
Abstract
Benchtop immobilized metal affinity chromatography (IMAC), of polyhistidine tagged proteins is easily mastered by undergraduate students and has become the most widely used protein purification method in the modern literature. But, the application of affinity chromatography to metal binding proteins, especially those with redox sensitive metals such as iron, is often limited to laboratories with access to a glove box – equipment that is not routinely available in the undergraduate laboratory. In this article, we demonstrate our benchtop methods for isolation, IMAC purification and metal-ion reconstitution of a poly-histidine tagged, redox-active, non-heme iron binding extradiol dioxygenase and the assay of the dioxygenase with varied substrate concentrations and saturating oxygen. These methods are executed by undergraduate students and implemented in the undergraduate teaching and research laboratory with instrumentation that is accessible and affordable at primarily undergraduate institutions.
Introduction
The first reports of the purification of a polyhistidine tagged protein from extracts of a host organism using chelation of the histidine tag by an immobilized metal entered the literature in 19881,2. Since that time, the addition of polyhistidine tags to recombinant proteins and their purification by immobilized metal affinity chromatography (IMAC) have become virtually ubiquitous in the biochemical literature3,4,5. IMAC purification methods can be implemented on the benchtop, using automated chromatography and in spin-column formats. While affinity purification methods, especially IMAC, are widely used in the research laboratory, they are less common in the undergraduate teaching laboratory. The most widely used laboratory textbooks for the biochemistry laboratory do not routinely teach these methods, instead opting for more traditional ion-exchange or dye-binding chromatography6,7,8,9. For example, the purification of lactate dehydrogenase by Anderson10 uses affinity by dye-binding, and the purification of Bovine milk α-lactalbumin7,11 by Boyer uses a nickel-nitriloacetic acid matrix, but no recombinant poly-histidine tag, relying instead on intrinsic affinity of the protein for the resin. Some modern undergraduate laboratory textbooks and publications do implement immobilized metal affinity chromatography on poly-histidine tagged protein targets such as green or red-fluorescent proteins12,13,14,15, antibodies16, and selected enzymes17,18,19,20, even some of unknown function21. Arguably, the purification of an enzyme is preferable in the teaching laboratory, because the target can be assayed for activity in subsequent sessions, enriching the experience of "real science" on the part of the student; indeed, these types of laboratory experiences have been published and beneficial outcomes on student learning reported17,18,20,21. And yet, applications of IMAC to enzyme purification in the biochemistry teaching laboratory remain sparse, and the published methods may even presume access to chromatography instrumentation that is typically unavailable for use in the classroom laboratory20. There are also limitations in the application of IMAC to metalloproteins, especially those which bind redox-sensitive divalent metals that are essential to activity22. Frequently, the metal ion is lost or oxidized during purification yielding an inactive enzyme ill-suited to the undergraduate laboratory.
A full one-third of enzymes bind a metal ion23, and despite a nearly universal requirement for iron in all forms of life23, iron is arguably among the most problematic metal ions in enzymology. Non-heme Fe2+ binding enzymes are particularly prone to loss and/or oxidation of the metal during IMAC; presumably due to the lack of a dedicated organic ligand like heme and the ease with which Fe2+ can dissociate from amino acid ligands24. Furthermore, the oxygen dependent oxidation of Fe2+ to Fe3+ is spontaneous in aqueous solution, due to the negative free energy change and the relative stability of Fe3+. Often, these challenges are overcome by use of anaerobic atmosphere and/or non-IMAC chromatographic methods22. In this article, we will demonstrate the use of benchtop IMAC to purify the Fe2+ dependent metalloenzyme L-DOPA dioxygenase using simple, inexpensive chromatography supplies, followed by the reconstitution of the active site Fe2+, and enzymatic assay. These methods are standard in our own undergraduate biochemistry laboratory of 6-12 student groups and can be used to expand the repertoire of enzyme investigations at the undergraduate level.
Protocol
1. Preparation for Purification
- Preparation of the cell-free crude extract
- Obtain a ~9-10 g cell pellet of E. coli (BL21) that overexpressed the polyhistidine tagged metalloprotein25 in a 50 mL conical tube.
- Add 5 mL per gram of room temp lysis/bind buffer (50 mM Phosphate, 300 mM NaCl, 10 mM imidazole at pH 8) to the ~9-10 g of cell pellet for a total volume ~45-50 mL. Periodically vortex to encourage dissolution. If the pellet was frozen, thaw in a tepid water bath.
- Using ice-water bath, chill the cell suspension to ~3 °C in preparation for cell lysis.
Note: At this point, cell lysis by sonication, bead milling or another method is possible. Bead milling is demonstrated here because it is rapid, relatively inexpensive, and does not require ear protection. - Assemble a 50 mL bead milling chamber according to the manufacturer’s instructions.
- Fill the bead mill chamber half full of chilled 0.1 mm glass beads that have been stored at -20 °C.
- Fill the rest of the chamber with the chilled cell suspension and use 4 °C lysis/bind buffer to fill the chamber completely.
- Use a small spatula to rotate the shaft of the bead mill and mix the cell suspension with the beads.
- Remove any large bubbles with a pipet, and then screw the lid on.
- Dry off the chamber from any leakage.
- Add about 1 cup of ice to the clear, plastic jacket, invert the filled 50 mL chamber into the ice filled jacket, and screw closed.
- Secure the ice-jacketed chamber onto the motor.
- Turn the bead mill motor on for 15 seconds at 15,800 rpm, then allow to rest for 45 seconds. Repeat 8 times.
- When the 8 cycles are complete, decant the entire cell lysis suspension into a 50 mL or larger tube rated for high speed centrifugation (25,000 x g or higher). Place a pair of balanced tubes in centrifuge and spin at 25,000 x g for 40-45 minutes at 4°C to pellet cell debris and glass beads.
Note: If 50 mL or larger tubes rated for high speed centrifugation are not available, remove the glass beads by centrifuging at 1000 – 2000 x g for 2-3 min in a 50 mL conical tube to yield a smaller volume of cell suspension (~25 mL) that can be decanted into a smaller volume tube rated for high speed centrifugation. - When centrifugation is complete, decant the clear, yellow, cell-free, crude extracts into a clean 50 mL conical tube. Take care not to transfer any cell debris.
- Collect a small sample of the cell-free crude extracts for later analysis of the purification by SDS-PAGE26.
- Preparation of the IMAC column
- Obtain a 1.5 x 20 cm column equipped with a lower bed support to retain resin particles, a Luer lock outlet and an upper cap that also contains a Luer lock fitting. Fit the Luer-lock outlet with a stopcock to control flow.
- Working at the benchtop, securely mount the column on a ring stand.
- Obtain a 50% slurry of nickel-bound nitriloacetic acid (Ni-NTA) resin in 20% ethanol that has been stored at 4 °C.
- Gently swirl the bottle to evenly re-suspend the resin.
- Working at room-temperature and on the benchtop, use a graduated pipet to withdraw 2 mL of the slurry, which will yield 1 mL of settled resin capable of binding 50-60 mg of polyhistidine tagged protein, and transfer the slurry to the column.
- Open the stopcock and allow the excess storage solution to drain by gravity from the resin.
- Close the stopcock securely
- Using a Pasteur pipet, carefully add chilled lysis/bind buffer (50 mM Phosphate, 300 mM NaCl, 10 mM imidazole at pH 8) and to the column of resin – at least 30 mL. Take care not to disturb the resin surface. Run the buffer down the walls of the column to prevent splashing.
- Equilibrate the resin by allowing the lysis/bind buffer to drain slowly from the column by gravity into a collection beaker.
- When the lysis/bind buffer has mostly drained, close the stopcock to stop the flow of buffer when ~5 mL of lysis buffer remains above the resin and leave the column, mounted upright, until ready to proceed or for up to 1 week.
2. Purification of Polyhistidine-tagged Target by IMAC
- Prepare wash buffer (50 mM Phosphate, 300 mM NaCl, 20 mM imidazole pH 8) and elution buffer (50 mM Phosphate, 300 mM NaCl, 250 mM imidazole, 10% glycerol pH 8). Chill at 4°C.
- Prepare the Ni-NTA column for the addition of the cell-free crude extracts by opening the stopcock and allowing remaining lysis/binding buffer to drain by gravity through the Ni-NTA resin. When all the buffer has drained, and the resin surface is exposed, close the stopcock.
- Using a Pasteur pipet, carefully pipet the cell free crude extracts onto the resin, taking care not to disturb the surface. Run the crude extracts down the walls of the column to prevent splashing. The volume of crude extracts applied is dependent on the level of expression of the polyhistidine-tagged protein; ensure the total volume of crude extracts contains 50-60 mg or less of the target protein per mL of Ni-NTA resin (see step 1.2.4).
- When all of the crude extracts have been transferred to the column, open the stopcock so the column can drain by gravity. This flow through is composed of proteins that did not bind to the resin – collect 100 μL for later analysis of the purification by SDS-PAGE.26
- The viscosity of the cell-free crude extracts can slow the gravity flow of the column considerably. To increase the rate of flow, apply a simple “hand-pump”:
- Take a 10 mL Luer-lock syringe and connect it to the cap of the column using a short length of plastic tubing and Luer-lock fittings between to the column cap and the syringe. Draw out the syringe plunger, and then tightly fit the column cap on the top of the column.
- Gently compress the plunger of the syringe while manually assessing the flow rate by collecting the eluent into a graduated cylinder; rates of 1-2 mL per minute are compatible with the resin and this setup. Gently increase compression of the syringe plunger as the flow rate slows.
- To prevent the introduction of air into the resin, remove the column cap and attached hand-pump when the meniscus of the applied liquid reaches 5-10 mm above the resin bed, and allow the remaining volume to drain by gravity.
- When the cell free crude extracts have finished draining and the resin surface is exposed again, use a Pasteur pipet to carefully add ~30 mL of chilled Wash buffer to the column – taking care not to disturb the surface of the resin. Run the buffer down the walls of the column to prevent splashing.
- Open the stopcock and allow the wash buffer to drain through the column. This process is removing weakly bound proteins from the resin. Discard the eluent.
- While the wash buffer is draining, prepare sixteen, labeled, microcentrifuge tubes for collecting 1 mL fractions.
- When the wash buffer has drained, and the resin surface is exposed again, close the stopcock. Use a Pasteur pipet to carefully add ~30 mL of chilled elution buffer to the column – taking care not to disturb the surface of the resin. Run the buffer down the walls of the column to prevent splashing.
- Open the stopcock slowly and allow the elution buffer to elute the polyhistidine tagged protein from the column. Collect 1 mL of eluent in each of the marked microcentrifuge tubes, 1 through 16.
- Test the fractions for protein using a colorimetric protein assay such as the Bradford assay or UV-Visible spectroscopy according to the methods specific by the reagent and/or instrument manufacturers27,28. As shown here, the colorimetric assay using Coomassie blue is scaled down to a 100 μL volume in order to sacrifice only a small volume from each fraction.
- Mix 3 μL of each fraction with 100 µL of a colorimetric protein assay reagent and manually observe the formation of any color to indicate the presence of protein in the fraction; detection with a spectrometer is not necessary.
- If protein is found in fraction 16, continue eluting 1 mL fractions and assay for protein periodically, until the eluted fractions no longer contain a significant amount of protein.
- Combine protein-containing fractions into a clean conical tube, and withdraw a 100 μL sample for later analysis by SDS-PAGE26.
- Proceed with reconstitution of the metal ion cofactor or freeze the protein in 3 mL aliquots at -80°C. Ensure that 10% glycerol is included in the elution buffer is a cryoprotectant to stabilize the pure protein during freezing.
- When elution of the Ni-NTA column is complete, that is, all the protein is off the column and collected in fractions, pass another 25 mL of elution buffer through the column, and store the column at 4 °C in ~5 mL of elution buffer for short-term storage, or lysis/bind buffer with 20% ethanol for longer-term storage.
3. Reconstitution of Target Protein with Fe2+
- Addition of Fe2+
Note: A non-heme iron (II) binding enzyme, such as the L-DOPA dioxygenase in this example, requires an Fe2+ ion for activity; however, the iron (II) readily oxidizes to iron (III), which is inactive, and a fraction of the protein purifies without any iron at all.- To begin the reconstitution of the metal, obtain 3 mL of the purified enzyme suspended in elution buffer. If frozen, thaw rapidly using a tepid water bath, and once thawed, put the protein on ice.
- Using the volume of protein in the tube, calculate the amount of sodium ascorbate (198.1 g/mol) necessary to make the final concentration 12.5 mM in the sample. Also, calculate the amount of dithiothreitol (DTT, 154.25 g/mol) necessary to make the final concentration 12.5 mM in the sample.
- Add the solid sodium ascorbate and DTT and gently, but thoroughly, mix to dissolve completely. Take care not to cause protein precipitation with overly aggressive mixing. Add a small amount of iron (II) sulfate heptahydrate (MW 278.01 g/mol) to the protein sample.
- In order to obtain a small enough quantity of the iron salt, tap a few 1 mm granules of FeSO4•7H2O solid out on a piece of weigh paper, fold the paper over the granule, and crush it with the flat side of a metal spatula.
- Add a grain of the resulting powder to the tube of protein.
- Vortex to mix and a rosy pink color will appear in the tube.
- Incubate the pink solution of protein, capped, for 10-30 minutes on ice. The pink color will slowly fade over time.
- Gel filtration
- Use a gel filtration column packed with 10 mL of spherical polyacrylamide gel with a molecular weight exclusion limit of approximately 6,000 and a hydrated particle size range of 90–180 µm in a 1.5 x 12 cm column, which is also equipped with a 10 mL reservoir. Ensure the column is equipped with lower and upper bed supports in the form of 30 um porous polyethylene disks to retain the resin in the column and prevent the resin bed from running dry. Mount the gel filtration column to a ring stand.
- If using a pre-packaged gel filtration column, remove the cap and pour off the excess buffer above the upper bed support.
- To equilibrate the column, begin by filling the reservoir with buffer compatible with subsequent assay and storage, for example, 50 mM NaH2PO4, 200 mM NaCl, 10% glycerol at pH 8.0.
- If using a pre-packaged gel filtration column, snap off the bottom tip of gel filtration column to start the flow of buffer and expose the Luer slip fitting. Fit a stopcock to the Luer slip end of the column, but leave it open and allow the column to drip by gravity.
- When the buffer has drained from the column and the upper bed support is exposed, close the stopcock, and add the ~3 mL of the Fe (II) reconstituted metalloenzyme to the column by pipetting the solution onto the exposed upper bed support.
- Open the stopcock and allow the entire 3.0 mL sample to enter the column by gravity. Discard these first 3 mL of eluent. Any pink color that forms while handling the solution will be trapped at the top of the column.
- When the column has stopped dripping and the upper bed support is exposed, add 4 mL of the gel-filtration buffer (e.g., 50 mM NaH2PO4, 200 mM NaCl, 10% glycerol at pH 8.0) to the column. Open the stopcock and collect the eluent into microcentrifuge tubes over eight 0.5 mL fractions.
- Test the fractions for protein using a colorimetric protein assay or UV-Vis27,28 as described earlier.
- Combine protein-containing fractions.
- Withdraw a sample for SDS-PAGE analysis26.
- Proceed with assay or storage of the sample.
Representative Results
These representative results were collected by undergraduate students as they executed this protocol during two course laboratory periods of BCM 341: Experimental Biochemistry at Muhlenberg College. Figure 1 demonstrates the results of the purification of a 20 kDa poly-histidine tagged metalloenzyme, L-DOPA dioxygenase, as performed by two undergraduate students during a 4-hour classroom laboratory period and analyzed by SDS-PAGE by the same students during a subsequent laboratory period. The poly-histidine tagged protein is effectively purified (Figure 1, lane 2). An immunoblot of the gel using antibody against the poly-histidine tag indicates that a small amount of the poly-histidine tagged target protein is lost in the flow-through (data not shown), likely because the greater than 100 mg quantity of target protein in the lysate exceeded the binding capacity of the resin.
Figure 2 demonstrates student-collected activity data on the enzymatic reaction of the poly-histidine tagged metalloenzyme target, L-DOPA dioxygenase, after reconstitution with iron (II) and subsequent gel-filtration as described by the protocol herein. The five second dead-time before data collected begins is typical of students executing this technique for the first time. The robust activity of the metalloenzyme was detected using a published assay, and yielded steady-state kinetic parameters consistent with published results29. Steady-state kinetic parameters were determined by fitting the progress curves30 shown in Figure 2; however, non-linear least squares fitting of initial rates collected over a series of substrate concentrations is equally possible31.
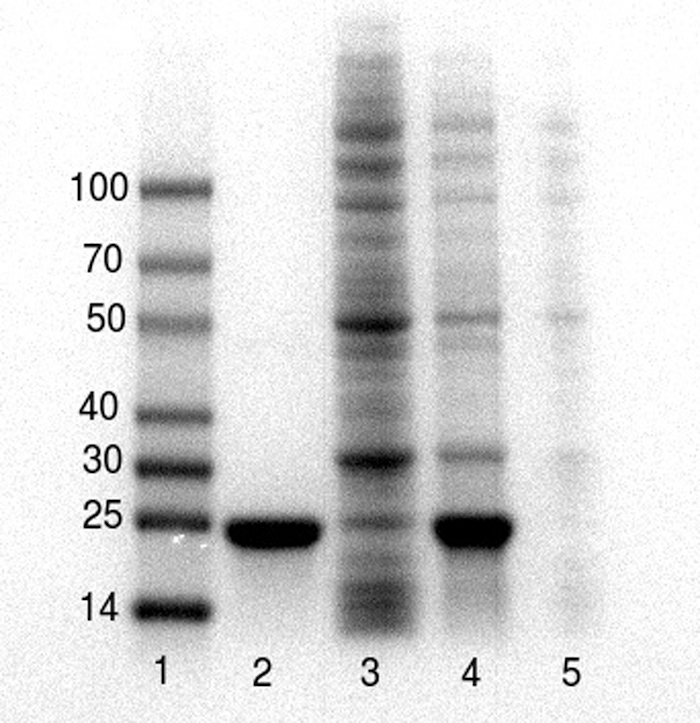
Figure 1. SDS-PAGE26 analysis of the poly-histidine tagged protein before, during and after purification. Lanes 1 – Molecular weight markers, 2-purified protein (20 kDa) post Ni-NTA, 3- Ni-NTA flow through, 4 – cell free crude extracts prior to purification, 5 – cell-debris pellet post lysis. Samples were prepared using 5x sample/Loading buffer and separated on pre-cast 4-20% polyacrylamide gels using a gel electrophoresis system. Please click here to view a larger version of this figure.

Figure 2. Steady state assay of the Iron (II) reconstituted metalloenzyme (i.e., L-DOPA dioxygenase, 10µM) with substrate (L-DOPA – 5 µM (red), 25 µM (green), 50 µM (blue)) in buffer (50 mM Phosphate, 200 mM NaCl, pH 8). Traces depict product formation at 414nm. Absorbance data were continuously acquired using 1 mL methacrylate cuvettes in a split-beam scanning UV-Visible spectrometer29. Raw data are fit to a model of the Michaelis-Menten steady-state approximation (KM 30.8 µM ± 14.4, kcatapparent 2.3 s-1 ± 0.05)30. Please click here to view a larger version of this figure.
Discussion
While the addition of polyhistidine tags to recombinant proteins and their purification by IMAC has become virtually ubiquitous in the biochemical literature3,4,5, applications of IMAC to enzyme purification in the biochemistry teaching laboratory remain sparse, and published methods do not always consider the resource limitations of the teaching laboratory20. Furthermore, the use of IMAC in the teaching laboratory is most effective when coupled to experiments that assess activity and purity, making IMAC purification of an enzyme an ideal instructional activity. In order to extend the application of IMAC to the purification of enzymes, including metalloenzymes, in the teaching laboratory, reliable and inexpensive methods are needed. In this protocol, we demonstrate benchtop IMAC using readily available and inexpensive laboratory supplies, while also addressing the limitations in the application of IMAC to metalloproteins22, by reconstituting the iron(II) dependent metalloenzyme, L-DOPA dioxygenase, post purification. Using the reagents and materials described , we estimate the cost of consumables for eight student groups is between $500-600 per semester to run this protocol, including the analysis steps outlined in Figure 1 and Figure 2.
Due to the ease with which Fe2+ can dissociate from amino acid ligands24 and the facile oxidation of Fe2+ by O2 to Fe3+, reconstitution of non-heme, amino acid-chelated iron(II) into a recombinant metalloenzyme is a typical component of the enzyme purification. When classical chromatography is used, it is possible to avoid total loss of iron in some cases32, but more often, the iron (II) is added back in the presence of reducing agents33,34,35,36 often under an anaerobic atomosphere37,38,39, and in some cases the excess iron is not removed33,34,36, complicating any subsequent assay. Consecutive steps of classical chromatography and an anaerobic atmosphere are not realistic for the undergraduate laboratory, prompting the development of this protocol.
While the manual preparation of the Ni-NTA column and the processing of samples largely by gravity does take additional time and effort when compared to pre-packaged columns and automated chromatography instrumentation, the manual steps allow for hands-on learning by the student that result in increased understanding of the science behind the process. The addition of an iron (II) salt under the conditions outlined here is particularly sensitive to excess dithiothreitol. If a student mistakenly adds an excess of dithiothreitol, a precipitation event is likely. We have found it helpful to require students to perform calculations of reagent quantities before arriving in lab, so laboratory time can be used most effectively at the bench. The entire benchtop IMAC purification – from cell-lysis to protein elution – can be accomplished in one 4-hour laboratory period, followed by reconstitution and assay in a subsequent lab period.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This publication is based upon work supported by the National Science Foundation under Grant No. CHE 1708237.
Materials
| consumables | |||
| BeadBeater 0.1mm glass beads | BioSpec Products | 11079101 | 1 pound each |
| 50mL Conical Tubes with Screw Caps, sterile | VWR | 21008-178 | |
| Sodium chloride | Fisher Bioreagents | BP358-1 | |
| Potassium phosphate, monobasic | Acros (Fisher) | AC42420-5000 | |
| Sodium Ascorbate | Acros (Fisher) | AC35268-1000 | |
| DTT (Dithiothreitol) | Lab Scientific | D-115 | |
| Iron(II) sulfate heptahydrate | Sigmaaldrich | 310077 | |
| HisPur NiNTA Resin | Fisher (pierce) | PI88221 | |
| Econo-Column Chromatography Columns – 1.5 x 20cm | Bio-Rad | 737-1522 | 1.5 x 20 cm, 35 ml, 2ea, |
| Stopcock Valve, one way, female to male luer | Kimble | 420163-0000 | pack of 50 |
| BD 10mL luer-loc syringe (non-sterile, without needle) | VWR | 301029 | |
| Fitting for tubing: 1.6 mm Barb to Female Luer | Biorad | 7318222 | |
| Fitting for tubing: 1.6 mm Barb to Male Luer | Biorad | 7318225 | |
| Silicon Tubing (1.6 mm ID/0.8 mm wall, for 0.2-5 ml/min on Peristaltic Pump) | Bio-Rad | 7318211 | Pkg of 1, 1.6 mm ID/0.8 mm wall, 10 m, low-pressure tubing for liquid handling |
| Glycerol | Fisher (Pierce) | 17904 | |
| Coomassie Plus (Bradford) Protein Assay | Thermo Scientitic | 23236 | |
| Microcentrifuge Tubes, snap cap, 1.5mL | VWR | 89000-028 | |
| Fisherbrand polystyrene disposable serological pipets | Fisher | 13-676-10F | |
| Fiserbrand universal pipet pump | Fisher | 14-955-110 | |
| Fisherbrand Transfer Pipets | Fisher | 13-711-9AM | |
| Econo-Pac 10DG desalting columns | Bio-Rad | 732-2010 | box of 30 |
| ExpressPlus PAGE 5x sample buffer | Genscript | MB01015 | 5mL (Dilute 1:5 with sample) |
| ExpressPlus PAGE Gel, 4-20%, 12 wells | Genscript | M42012 | 20 gels |
| Fisherbrand Disposable Cuvettes, Methyacrylate | Fisher | 14-955-128 | case of 500 |
| Cuvette Caps Square Disposable | Fisher | 14-385-999 | |
| L-DOPA (3,4-dihydroxyphenylalanine) | Acros | D9628-5G | |
| Permanent Equipment: | |||
| BeadBeater 50mL chamber | BioSpec Products | 110803-50SS | 1 chamber |
| BeadBeater | BioSpec Products | 1107900-101 | 350 ml polycarbonate chamber, rotor assembly, motor base, ice-water cooling jacket and one pound of glass beads. |
| Centrifuge tubes, High-Speed PPCO, 50mL | Fisher | 3119-0050PK | |
| Mini-PROTEAN Tetra Vertical Electrophoresis Cell for Mini Precast Gels, 4-gel | Bio-Rad | 1658004 | 4-gel vertical electrophoresis system, includes electrode assembly, companion running module, tank, lid with power cables, mini cell buffer dam |
| PowerPac HC Power Supply | Bio-Rad | 1645052 | 100–120/220–240 V, power supply for high-current applications, includes power cord |
| UV-1800 with UV-Probe Software | Shimadzu | UV-1800 | |
| Kintek Global Kinetic Explorer | Kintek Corp | version 6 | https://www.kintekexplorer.com/downloads/ |
References
- Hochuli, E., Bannwarth, W., Döbeli, H., Gentz, R., Stüber, D. Genetic Approach to Facilitate Purification of Recombinant Proteins with a Novel Metal Chelate Adsorbent. Nature Biotechnology. 6 (11), 1321 (1988).
- Smith, M. C., Furman, T. C., Ingolia, T. D., Pidgeon, C. Chelating peptide-immobilized metal ion affinity chromatography. A new concept in affinity chromatography for recombinant proteins. Journal of Biological Chemistry. 263 (15), 7211-7215 (1988).
- Block, H., et al. Chapter 27 Immobilized-Metal Affinity Chromatography (IMAC): A Review. Methods in Enzymology. , 439-473 (2009).
- Derewenda, Z. S. The use of recombinant methods and molecular engineering in protein crystallization. Methods. 34 (3), 354-363 (2004).
- Gräslund, S., et al. Protein production and purification. Nature Methods. 5 (2), 135-146 (2008).
- Switzer, R. L., Garrity, L. F. . Experimental Biochemistry. , (1999).
- Boyer, R. . Modern Experimental Biochemistry, 3rd Edition. , (2001).
- le Maire, M., Chabaud, R., Hervé, G. . Laboratory Guide to Biochemistry, Enzymology, and Protein Physical Chemistry: A Study of Aspartate Transcarbamylase. , (2012).
- Bettelheim, F. A., Landesburg, J. M. . Laboratory Experiments for General, Organic, and Biochemistry. , (2013).
- Anderson, A. J. Affinity chromatography of lactate dehydrogenase: An experiment for the undergraduate biochemistry laboratory. Journal of Chemical Education. 65 (10), 901 (1988).
- Boyer, R. F. Purification of milk whey α-lactalbumin by immobilized metal-ion affinity chromatography. Journal of Chemical Education. 68 (5), 430 (1991).
- Moffet, D. A. From gene mutation to protein characterization. Biochemistry and Molecular Biology Education. 37 (2), 110-115 (2009).
- Sommer, C. A., Silva, F. H., Novo, M. T. M. Teaching molecular biology to undergraduate biology students: An illustration of protein expression and purification*. Biochemistry and Molecular Biology Education. 32 (1), 7-10 (2004).
- Wu, Y., Zhou, Y., Song, J., Hu, X., Ding, Y., Zhang, Z. Using green and red fluorescent proteins to teach protein expression, purification, and crystallization. Biochemistry and Molecular Biology Education. 36 (1), 43-54 (2008).
- Ward, W. W., Swiatek, G. C., Gonzalez, D. G. Green fluorescent protein in biotechnology education. Methods in enzymology. 305, 672-680 (2000).
- Kay, B. K., Winter, J., McCafferty, J. . Phage Display of Peptides and Proteins: A Laboratory Manual. , (1996).
- Arkus, K. A. J., Jez, J. M. An integrated protein chemistry laboratory. Biochemistry and Molecular Biology Education. 36 (2), 125-128 (2008).
- Crowley, T. E. Expression, purification, and characterization of a recombinant flavin reductase from the luminescent marine bacterium Photobacterium leiognathi. Biochemistry and Molecular Biology Education. 38 (3), 151-160 (2010).
- Colabroy, K. L. A writing-intensive, methods-based laboratory course for undergraduates. Biochemistry and Molecular Biology Education: A Bimonthly Publication of the International Union of Biochemistry and Molecular Biology. 39 (3), 196-203 (2011).
- Kreiling, J. L., Brader, K., Kolar, C., Borgstahl, G. E. A real-time and hands-on research course in protein purification and characterization: Purification and crystal growth of human inosine triphosphate pyrophosphatase. Biochemistry and Molecular Biology Education. 39 (1), 28-37 (2011).
- Gray, C., et al. Known structure, unknown function: An inquiry-based undergraduate biochemistry laboratory course. Biochemistry and Molecular Biology Education. 43 (4), 245-262 (2015).
- Kocabas, E., Hernick, M. Metalloenzymes: Use of Recombinant Protein Expression and Affinity Tags to Aid Identification of Native Metal Ion Cofactors. Biochemistry & Analytical Biochemistry. 2 (2), 1-3 (2013).
- Ellis, W. R. Metalloenzymes. Reviews in Cell Biology and Molecular Medicine. , (2006).
- Williams, R. J. P., Begley, T. P. Metallo-Enzymes and Metallo-Proteins, Chemistry of. Wiley Encyclopedia of Chemical Biology. , (2007).
- Mierendorf, R. C., Morris, B. B., Hammer, B., Novy, R. E. Expression and Purification of Recombinant Proteins Using the pET System. Molecular Diagnosis of Infectious Diseases. , 257-292 (1998).
- He, F. Laemmli-SDS-PAGE. BIO-PROTOCOL. 1 (11), (2011).
- He, F. Bradford Protein Assay. BIO-PROTOCOL. 1 (6), (2011).
- Johnson, M. Protein Quantitation. Materials and Methods. , (2017).
- Colabroy, K. L., Hackett, W. T., Markham, A. J., Rosenberg, J., Cohen, D. E., Jacobson, A. Biochemical characterization of L-DOPA 2,3-dioxygenase, a single-domain type I extradiol dioxygenase from lincomycin biosynthesis. Arch Biochem Biophys. 479 (2), 131-138 (2008).
- Johnson, K. A. Fitting enzyme kinetic data with KinTek Global Kinetic Explorer. Methods in Enzymology. , 601-626 (2009).
- Kemmer, G., Keller, S. Nonlinear least-squares data fitting in Excel spreadsheets. Nature Protocols. 5 (2), 267-281 (2010).
- Wang, Y. Z., Lipscomb, J. D. Cloning, overexpression, and mutagenesis of the gene for homoprotocatechuate 2,3-dioxygenase from Brevibacterium fuscum. Protein Expr Purif. 10 (1), 1-9 (1997).
- Amaya, A. A., Brzezinski, K. T., Farrington, N., Moran, G. R. Kinetic analysis of human homogentisate 1,2-dioxygenase. Archives of Biochemistry and Biophysics. 421 (1), 135-142 (2004).
- Johnson-Winters, K., Purpero, V. M., Kavana, M., Nelson, T., Moran, G. R. 4-Hydroxyphenyl)pyruvate Dioxygenase from Streptomyces avermitilis: The Basis for Ordered Substrate Addition. Biochemistry. 42 (7), 2072-2080 (2003).
- Bugg, T. D. H. Overproduction, purification and properties of 2,3-dihydroxyphenylpropionate 1,2-dioxygenase from Escherichiacoli. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1202 (2), 258-264 (1993).
- Mendel, S., Arndt, A., Bugg, T. D. H. Acid-Base Catalysis in the Extradiol Catechol Dioxygenase Reaction Mechanism Site-Directed Mutagenesis of His-115 and His-179 in Escherichia coli 2,3-Dihydroxyphenylpropionate 1,2-Dioxygenase (MhpB). Biochemistry. 43 (42), 13390-13396 (2004).
- Viggiani, A., Siani, L., Notomista, E., Birolo, L., Pucci, P., Di Donato, A. The Role of the Conserved Residues His-246, His-199, and Tyr-255 in the Catalysis of Catechol 2,3-Dioxygenase from Pseudomonas stutzeri OX1. Journal of Biological Chemistry. 279 (47), 48630-48639 (2004).
- Uragami, Y., et al. Crystal structures of substrate free and complex forms of reactivated BphC, an extradiol type ring-cleavage dioxygenase. Journal of Inorganic Biochemistry. 83 (4), 269-279 (2001).
- Veldhuizen, E. J. A., et al. Steady-state kinetics and inhibition of anaerobically purified human homogentisate 1,2-dioxygenase. Biochemical Journal. 386 (Pt 2), 305-314 (2005).
