Figures 1 and 2 show how the laser pointer and laser goggles can be used to quickly and inexpensively genotype GFP positive animals from a litter. In cases of young mouse pups, the technique can be used to noninvasively identify GFP label in the animal's brains through the skull and overlying skin (Figure 1A – D). Emitted fluorescence can be seen through the skin and the skull of mouse pups at least up to postnatal day 3 (Figure 1C). The procedure is shown in Figure 1 for our green-fluorescent protein (GFP) labeled mouse line expressing GFP in its glycinergic subpopulation of neurons (GlyT2-GFP). In older animals with thicker skulls and pigmented skin, the GFP label shows less well through skin and bone, and therefore the skin on the head and/or neck needs to be removed before the optical genotyping (Figure 2A).
After perfusing the animal the brain is taken out and illuminated again to find the fluorescently labeled nuclei of interest. The bright structures that light up close to the ventral surface of the brainstem in Figure 2B are the two ventral nuclei of the trapezoid body, which we used as both a guide and a target for our tracer injections. Figures 4 and 5 show representative results of an anterograde injection into the ventral nucleus of the trapezoid body (VNTB) using tetramethylrhodamine dextrane (TRITC; Figure. 4) and two retrograde injections into the medial nucleus of the trapezoid body (MNTB) using choleratoxin subunit-b (CTB; Figures 5A and B) and TRITC (Figures 5C and D). Bright axonal labeling indicating (in this case inhibitory) projections from VNTB to MNTB can be seen following an anterograde injection into VNTB (Figure 4). Figures 5A and 5C show an overview over the site of injection (MNTB) with the nucleus containing the retrogradely labeled cells (VNTB) next to it. Figures 5B and 5D show magnified images of retrogradely labeled somata of glycinergic VNTB cells in the same sections as displayed in Figure 5A and 5C. Note how the retrograde tracing with CTB results in a more punctate pattern12-13 (Figure 5B), whereas VNTB cells retrogradely labeled with TRITC display a very dense Golgi-like labeling21 (Figure 5D).
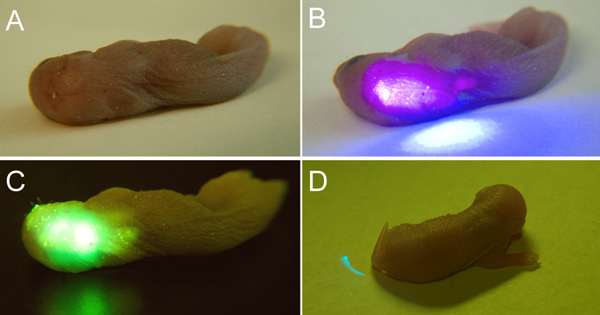
Figure 1: Optical Genotyping of GlyT2-GFP-positive and GlyT2-GFP-negative Mice. The GFP-positive mice were photographed under (A) normal lighting conditions, (B) upon laser illumination with a 405 nm laser pointer without the green-filtering goggles and (C) upon laser illumination with the laser and UV safety goggles. A strong GFP signal is visible in the area above the cerebellum and the brainstem. (D) By contrast, GlyT2-GFP-negative pups from the same mouse line and age (p2) do not show any signs of fluorescence in the same areas (back of the head and spinal cord). The blue laser light is effectively filtered by the goggles.
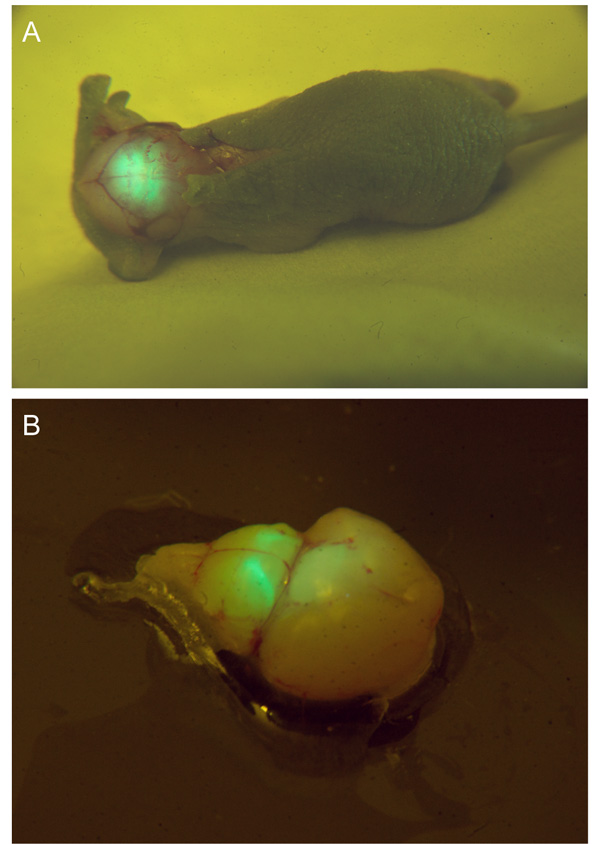
Figure 2: Optical Genotyping and a Prepared Brain Explant. (A) A p4 GlyT2-GFP mouse pup's skull is exposed to laser illumination and viewed through band-pass filtering goggles. The fluorescing GFP can be observed through the skull. (B) A brain explant from the same pup in a preparation dish upon laser illumination, as seen through the filtering goggles. Glycinergic axons and brain nuclei, such as the ventral nucleus of the trapezoid body (VNTB) can be seen as very bright structures close to the brain surface.

Figure 3: Overview Over an In Vitro Pressure Injection and Electroporation Setup. (A) 1: stereoscope, 2: mounted laser pointer (405 nm wavelength), 3: headstage with injection pipette mounted on a micromanipulator, 4: preparation dish containing the brain explant, 5: a PC with installed MC Stimulus software, 6: pressure injection device, connected to the injection pipette via tubing. (B) 7: stimulus isolation unit, 8: high-intensity illuminator, 9: stimulator. The stimulator is connected to the electrode in the pipette via the stimulation isolation unit and is driven by the PC.
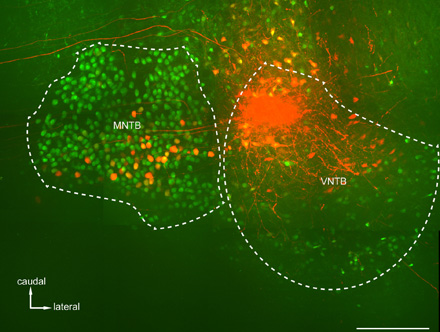
Figure 4: Anterograde Tracing of Inhibitory Connections from VNTB to the Medial Nucleus of the Trapezoid Body (MNTB). Shown is a maximum projection of a confocal stack taken in a horizontal slice spanning about 200 microns in depth in a cleared brain of a p14 GlyT2-GFP mouse. Following a tetramethylrhodamine dextrane (TRITC) injection into the caudo-medial portion of the VNTB, brightly labeled axonal connections (and in some cases their terminal endings) from VNTB to MNTB can be observed. Scale bar: 200 μm.
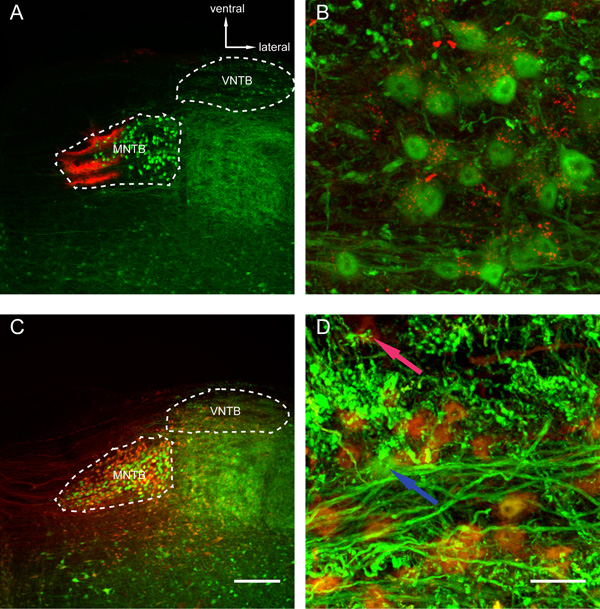
Figure 5: Retrograde Tracer Injections into MNTB Reveal Filled Glycinergic Cells in the VNTB. Shown are maximum projections (spanning over approx. 50 μm) of confocal stacks taken in coronal slices of a p88 (A, B) and a p80 (C, D) GlyT2-GFP mouse. (A) An overview of a choleratoxin subunit-b (CTB) injection into the medial portion of the MNTB. (B) Glycinergic cells in the VNTB are filled with CTB (red puncta inside the cell somata) as a result of the injection shown in Figure 5A. Some of the puncta appear outside of the cells, which could be indicative of other GFP-negative (non-glycinergic) cell types in the VNTB being filled, as well (because of injured fibers of passage in the area of injection, for example). (C) Another overview of a retrograde TRITC injection targeting the MNTB. Note, how a number of cells is filled with TRITC in the MNTB following electroporation (as opposed to the purely pressure-injected CTB in Figure 5A). (D) Retrograde labeling of glycinergic VNTB cells after a TRITC injection in the MNTB (same injection as shown in Figure 5C). Most of the cells display a dense yellow or orange labeling, because of the mixture of two fluorescent signals (the indigenous GFP expression and the filling with the red TRITC). Compare with a GFP-positive cell that for some reason did not take up the dye (blue arrow) and a tracer-labeled GFP-negative cell appearing deeply red and possibly filled, because of an axonal injury in the area of injection (red arrow). Note the different staining pattern of cell somata in retrograde tracer injections using CTB (Figure 5B) and TRITC (Figure 5D). Scale bars: Figures 5A and 5C: 200 μm, Figures 5B and 5D: 20 μm.
