This report describes the complete protocol for the design and construction of ESFs and splicing reporters. It also outlines the further application of ESFs in manipulating the AS of endogenous genes16. To illustrate typical results of ESF-mediated splicing changes, we use the data from our previous work as an example. The ESFs with different functional domains can be used to promote or inhibit the inclusion of the target cassette exon (Figure 1D & E). ESFs can also affect the usage of alternative 5' and 3' splice sites in the reporter system (Figure 1F & G).
The alternative splicing of the endogenous gene can also be specifically regulated with designer ESFs. We have demonstrated this application by specifically targeting Bcl-x, which can be spliced into two antagonistic isoforms with alternative 5' splice sites. We designed an ESF, Gly-PUF(531), that recognizes an 8-nt RNA element between the alternative 5' splice sites. This Gly-PUF(531) specifically shifted the splicing towards the production of Bcl-xS (Figure 2A). After transfecting the Gly-PUF(531) into HeLa cells, the level of Bcl-xS isoforms and Bcl-xS proteins increased in a dose-dependent manner, whereas the control ESF, Gly-PUF(WT), did not affect the ratio of Bcl-xS to Bcl-xL (Figure 2B & C). In addition, the designer ESF can induce the cleavage of caspase 3 and poly (ADP-ribose) polymerase (PARP), two well-known molecular markers of apoptosis (Figure 2D). As expected, the designer ESFs are predominantly localized in the nuclei of transfected cells, as demonstrated by immunofluorescence microscopy (Figure 2E). Consistently, the splicing shift by Gly-PUF(531) caused the fragmentation of nuclear DNA, indicating that these cells are undergoing apoptosis (Figure 2E). The increase of apoptotic cells was further confirmed by examining more than 200 cells from randomly selected fields and by quantifying the percent of cells with fragmented nuclear DNA (Figure 2F).
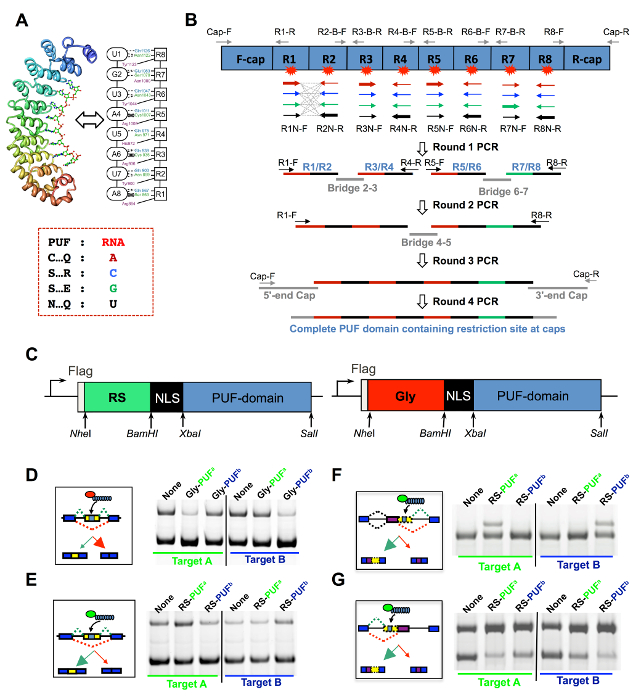
Figure 1: Design of ESFs and Their Activity in Modulating Exon Skipping. (A) Specific binding between the PUF domain and RNA targets is illustrated with the RNA-PUF structure and a schematic diagram. The PUF binding code for each of the four RNA bases, shown on the right with different colors, is used to design PUF mutations. (B) Flow chart to obtain a customized PUF domain. The PUF that recognizes "UGUAUAUA" was used as an example. A 4-round PCR strategy is used to assemble a PUF scaffold with customized RNA-binding specificity (color coded similarly to panel A). In the first round, a series of PCR primers that incorporate the desired RNA-recognition codes for two adjacent PUF repeats are used to generate four fragments that include the eight RNA-recognition codes of a full PUF protein (R1/R2, R3/R4, R5/R6, and R7/R8). Cap fragments encoding an N-terminal nuclear localization signal, a C-terminal stop codon, and bridge fragments are also produced separately (5'-end and 3'-end cap, bridge 2/3, bridge 4/5, and bridge 6/7). In the second round, new templates are generated by mixing overlapping fragments encoding adjacent repeats with the appropriate bridge (e.g., mixing R1/R2, R3/R4, and bridge 2/3 generates the template for R1 – 4) and then extending with DNA polymerase to fill the gaps. Similarly, the third round joins R1-4, R5-8, and bridge 4/5. Finally, the fourth round adds the 5'-end and 3'-end caps of the PUF domain together with the cloning sites for subsequent cloning to expression vectors. (C) Modular domain organization of ESFs. ESFs are driven by CMV promoters (arrow) and encode, from the N- to the C-terminal: a FLAG epitope (for the detection of ESFs), a functional module (a Gly-rich domain or an RS domain), an NLS (facilitating the nuclear localization of ESFs), and an RNA-recognition domain (a PUF domain). NheI and BamHI are designed to insert a functional module, while XbaI and SalI are designed to insert an RNA-recognition domain. (D) Gly-PUF ESFs are co-expressed with exon skipping reporters, and the splicing pattern is assayed by RT-PCR. The modified PUFa and PUFb specifically bind to 8-mer targets A and B, respectively (in the same colors). All combinations are used, so the PUF-target pairs of different color serve as the controls. The effects of RS-PUF on exon skipping (E), the competing 5' splice site (F), or the competing 3' splice site reporter (G) were assayed by methods similar to panel D. The data of the RT-PCR are from Wang et al.16. Please click here to view a larger version of this figure.
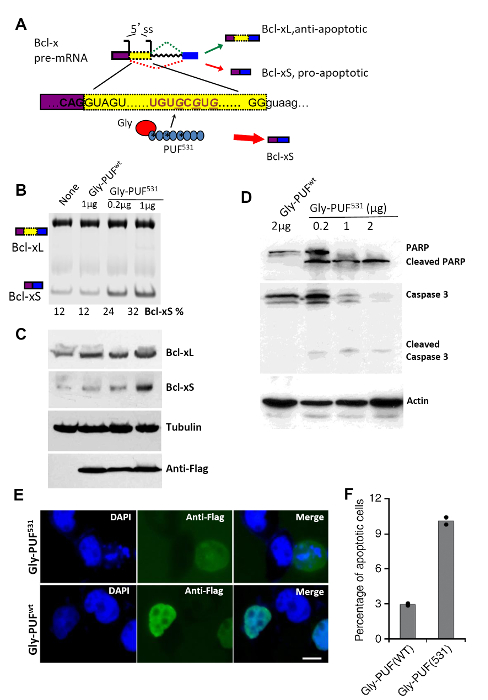
Figure 2: Regulation of Endogenous Bcl-x pre-mRNA Splicing with ESFs. (A) Schematic of the alternative splicing of endogenous Bcl-x pre-mRNA. Two alternative 5' splice site in exon 2 of Bcl-x are used to generate two isoforms of different sizes, Bcl-xL and Bcl-xS. The sequence UGUGCGUG between the two 5' splice sites is selected as the ESF target, and WT PUF repeats 1, 3, and 5 (Q867E/Q939E/C935S/Q1011E/C1007S) are reprogrammed (asterisks) to recognize this target sequence. The resulting ESF containing a Gly-rich domain inhibits the use of the downstream 5' ss (indicated by the red arrow). (B) Modulation of Bcl-x 5' ss usage. Different amounts of the Gly-PUF(531) expression construct are transfected into HeLa cells. Gly-PUF(WT) is used as a control. Two isoforms of Bcl-x are detected with RT-PCR using primers corresponding to exons 1 and 3 of the Bcl-x gene. The percentage of the Bcl-xS isoform is quantified and shown at the bottom. (C) ESFs affect the expression levels of Bcl-xL and Bcl-xS. Samples are loaded in the same order as in panel B, and all proteins are detected by Western blots. The expression of ESFs is detected by the anti-FLAG antibody, and the tubulin level is used as a control. (D) Different amounts of ESF expression constructs are transfected into HeLa cells, resulting in the cleavage of PARP and caspase 3. Samples are detected by Western blot 24 h after transfection. The actin level is detected as a control. (E) The subcellular localization of ESFs in transfected HeLa cells is detected by immunofluorescence microscopy with the anti-FLAG antibody. The cells are co-stained with DAPI to show the nuclei. Some nuclei, especially in cells transfected with Gly-PUF(531), are fragmented due to apoptosis. Scale bar: 5 µm. (F) Percentage of apoptotic cells (i.e. cells with fragmented nuclear DNA) are measured from randomly chosen fields of fluorescence microscopy images. The bars indicate the mean, while the dots indicate the data from the two experiments. The figures are modified from our earlier report by Wang et al.16 in accordance with the policy of Nature Publishing Group. Please click here to view a larger version of this figure.
