Experimental Procedure :
The assay requires synthetic peptides and anti-peptide antibodies. Selected peptides should be unique to the protein of interest, contain between 8 and 22 amino acids, and have no known post-translational modifications. Methionine residues are generally avoided and peptides containing dibasic amino acids (e.g. KK, KR, RR) are undesirable. For this technique, it is common to use stable isotope labeled peptides as internal standards, incorporating heavy (13C and 15N) labeled amino acids at the C-terminus of the peptide (i.e. K or R labeled).
The following protocol describes an assay developed to measure the peptide GDSLAYGLR from the mouse protein Osteopontin, using anti-peptide antibodies obtained from Epitomics Inc. (Burlingame, CA) and synthetic peptides from New England Peptide (Gardner, MA). The protocol consists of three main steps (Figure 1): 1) Trypsin digestion of the complex protein mixture, 2) Enrichment of peptides 3) Analysis by mass spectrometry. It will be demonstrated on a human plasma sample spiked with the mouse Osteopontin protein.
1. Trypsin enzymatic digestion and cleanup
- Thaw 10 μL neat plasma aliquot on wet ice.
- Determine the total protein concentration by BCA assay and centrifuge the sample to remove any suspended solids.
- Pipet 10 μL aliquot from its storage tube to a 1000 μL deepwell plate and cover with pierce-able film.
- Add 20 μL of fresh 9M urea / 30mM dithiothreitol (DTT) (final concentration 6M urea / 20mM DTT) to each sample.
- Incubate for 30 minutes at 37°C.
- Add 3 μL of fresh 500 mM iodoacetamide (final IAM 50mM) to each sample.
- Incubate for 30 minutes in the dark at room temperature.
- Add 257 μL of 100 mM Tris (pH 8) (dilutes urea to ˜0.6M).
- Add 10 μL of trypsin stock solution (1 μg/μL; for 1:50 enzyme:substrate ratio).
- Incubate 37°C overnight (12-16 hours).
- Add 3 μL of neat formic acid (final concentration of 1%).
- Add stable isotope standard (multiple standards are added if performing a multiplexed assay, typically this is about 10 μL containing 50-100 fmol of standard isotopically-labeled peptide).
- Wash the Oasis cartridge plate well with 500 μL of 0.1% formic acid in 80% acetonitrile, discarding the flow-through. Repeat this 3 times.
- Equilibrate the cartridge plate by adding 500 μL of 0.1% formic acid in water, and discard the flow-through. Repeat this 4 times.
- Load digest samples to the cartridge plate and adjust the vacuum so the flow is very slow.
- Wash with 500 μL of 0.1% formic acid in water, and discard flow-through. Repeat this 3 times.
- Elute peptides by adding 2 x 500 μL of 0.1% formic acid in 80% acetonitrile into 1000 μl deep-well plate (do not discard the flow-through).
- Lyophilize (or speedvac) the eluate to dryness. (Lyophilization is the preferred method)
- Reconstitute dried peptides by adding 50 μL PBS + 0.03% CHAPS.
2. Peptide immunoaffinity enrichment
- Transfer the sample to standard Kingfisher 96 well plates.
- Add 1 μg antibody and 1.5 μL Protein-G coated magnetic beads per target (it is optional to crosslink the antibody to the beads prior to addition). Ensure beads are well suspended by shaking or vortexing.
- Cover the plate with foil.
- Incubate overnight (12-16 hours) with gentle tumbling to ensure beads are suspended.
- Centrifuge plate at 32 x g for 5 seconds to remove any liquid from the foil surface.
- Remove the foil cover.
- Washing and elution of the beads can be performed manually or in an automated fashion. This procedure describes the steps automated on a Kingfisher platform.
- Wash the beads 2 times with 200 μL PBS + 0.03% CHAPS (1 minute per wash).
- Wash the beads 1 time with 200 μL 1:10 dilution of PBS + 0.03% CHAPS (1 minute).
- The peptides are eluted in 25 uL of 5% acetic acid + 0.03% CHAPS.
- Place the elution plate on a magnet and transfer the eluate to a 96 well plate, taking care not to transfer the leftover beads.
- Cover the plate with a sealing mat.
- The plate containing the eluate is delivered to the triple quadrapole mass spectrometer for analysis.
3. Analysis by multiple reaction monitoring – mass spectrometry
- Transitions for SRM/MRM analysis can be selected and optimized by infusing a 1 picomole per microliter solution of peptide standard in 30% acetonitrile/0.1% formic acid into the mass spectrometer at 0.5 μL/min. Once stable spray is obtained, collect an MS/MS spectrum.
- Transitions are selected from the MS/MS spectrum by identifying three abundant fragment ions in a region of the spectrum with little noise. For multiply-charged peptide ions, the y-ion fragments detected at m/z > precursor m/z are typically the best for SRM/MRM assay development. Figure 2 shows the MS/MS spectrum and selected transition ions 476.3 > 508.3, 579.3, 692.4 (transitions for the heavy stable isotope standard 481.3 > 518.3, 589.3, 702.4 not shown) for the peptide GDSLAYGLR.
- Depending on the make and model of the mass spectrometer used, there are multiple parameters that can be optimized for each transition. Here, we optimized the collision energy of each selected transition by ramping the collision energy and monitoring the level of signal. A collision energy of 25 was used for each transition.
- Briefly, a typical configuration for SRM/MRM analysis is as follows: Mobile phase (A) 0.1% Formic acid; (B) 90% Acetonitrile / 0.1% Formic acid, 0.3 x 5 mm C18 trap column, 75 μm ID x 15 cm C18 analytical column (Reprosil-Pur C18 AQ, 120 A° pore). The injection volume is 10 μL and the sample is loaded for 5 min at 3% B at a total flow rate 3 μL/min and eluted by a linear gradient from 3 – 45 %B at 300 – 400 nL/min in 10 min. Conditions on a 4000 QTRAP (ABSCIEX, Foster City, CA) were spray voltage 2.3kV, ion source temperature 150 °C, GS1 of 12, and curtain gas 15.
- The sample is analyzed by SRM/MRM by injecting 10 μL of the sample. Peak areas are integrated for light and heavy peptides using the program Skyline.14 Calculate the peak area ratio (PAR) of unlabeled/labeled peptide in each sample using the most abundant transition that is free of background noise, in this case, the y5 transition (light peptide 476.3 > 579.3, heavy standard peptide 481.3 > 589.3).
4. Representative Results:
Measured peak area ratios (light endogenous peptide relative to spiked heavy isotopically-labeled peptide) provide a quantitative measure of the target peptide. Figure 3 shows an example chromatogram of light and heavy peptides in a SISCAPA-enriched sample. Note that light and heavy peptides elute at the same time and multiple transitions can be monitored for each peptide to confirm the identity.
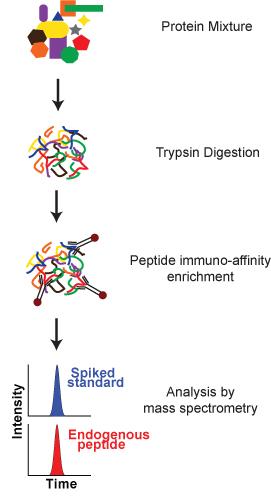
Figure 1. A schematic overview of the SISCAPA process. A complex protein mixture is digested into peptides. Targeted peptide analytes (endogenous analyte and a spiked stable isotope-labeled internal standard) are enriched using anti-peptide antibodies immobilized on Protein G-coated magnetic particles. Following isolation, the targeted peptides are eluted from the magnetic particles and analyzed by mass spectrometry for quantitation relative to the internal standard.
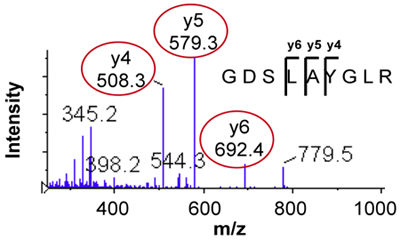
Figure 2. MS/MS spectrum of the peptide GDSLAYGLR showing selection of three fragments for SRM/MRM transitions.
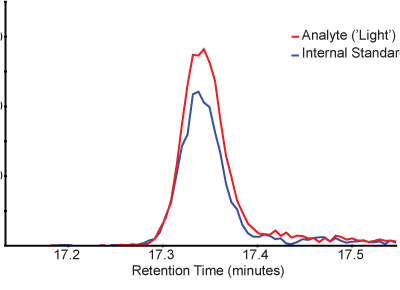
Figure 3. Example chromatograms showing the peak profile of a light peptide analyte (red) and the heavy stable isotope-labeled internal standard (blue). Chromatograms for the y5 transition from the light peptide (476.3 > 579.3) and heavy stable isotope labeled standard (481.3 > 589.3) are plotted over time as they elute from the chromatography system.
