1. Cell culture (Figure 1)
- Grow cells in minimal medium with trace elements, salts, vitamins, and specifically labeled carbon substrates that are best for pathway investigation. Use either shaking flasks or bioreactors for cell culture. Organic nutrients, such as yeast extract, may interfere with the measurement of amino acid labeling and thus cannot be present in the culture medium.
- Monitor cell growth by the optical density of the culture at an optimal wavelength (e.g., OD730 for Cyanothece 51142) with a UV/Vis spectrophotometer.
- Cells can first be grown in a non-labeled medium. The middle-log growth phase cells are preferred to be used for inoculation (3% (v/v) by volume inoculation ratio) of the labeled medium. The labeled culture should be sub-cultured (3% v/v inoculation ratio) in the same labeled medium to avoid the introduction of non-labeled carbon from the initial inoculum.
2. Amino acid extraction
- Harvest sub-cultured cells (10mL) in the middle-log growth phase by centrifugation (10 min, 8000×g).
- Resuspend the pellet in 1.5mL of 6M HCl and transfer it to a clear glass, screw-top GC vial. Cap the vials and place them in a 100°C oven for 24 hours to hydrolyze the biomass proteins into amino acids. Hydrolysis of biomass pellets can yield 16 of the 20 common amino acids (Figure 2) 5. Cysteine and tryptophan are degraded, and glutamine and asparagine are converted to glutamate and aspartate, respectively.
- Centrifuge the amino acid solution at 20,000×g for 5 min using 2 ml Eppendorf tubes, and transfer the supernatants to new GC vials. This step removes solid particles in the hydrolysis solution.
- Remove the GC vial lids and dry the samples completely under a stream of air using a Thermo Scientific Reacti-Vap evaporator (note: a freeze dryer can also be used to dry samples). This step can be done overnight.
3. Amino acid derivatization and GC-MS conditions
Analysis of amino acids or charged/highly polar metabolites via GC requires that these metabolites be derivatized, so that the amino acids are volatile and can be separated by gas chromatography 2.
- Dissolve the dried samples with 150 μL of tetrahydrofuran (THF) and 150 μL of N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide derivatization reagent.
- Incubate all samples in an oven or a water bath between 65 and 80°C for 1 hour. Vortex occasionally to make sure the metabolites in the vial are dissolved.
- Centrifuge the samples at 20,000×g for 10 min, and then transfer the supernatant to new GC vials. The supernatant should be a clear and yellowish solution. Due to saturation of the detectors, GC-MS measurement accuracy can be affected by the high concentration of injected TBDMS derivatized amino acids (these samples often shows dark brown color), therefore, we should dilute these samples using THF before GC-MS measurement 6.
- Analyze the samples by GC-MS (use a 1:5 or 1:10 split ratio, injection volume = 1 μL, carrier gas helium = 1.2 mL/min). Use the following GC temperature program: hold at 150°C for 2 minutes, increase at 3°C per min to 280°C, increase at 20°C per min to 300°C, and then hold for 5 minutes. Solvent delay can be set as ~5 min (for a 30 meter GC column). The range of the mass to charge ratio (m/z) in MS can be set between 60 and 500.
4. GC-MS data analysis
- TBDMS derivatized amino acid measurement can also be affected by isotope discrimination in GC separation. Light isotopes move slightly faster than heave isotopes in GC column. To reduce the potential measurement bias, we may average the mass spectrum of the whole amino acid peak range 6
- The GC and MS spectra of TBDMS derivatized metabolites have been reported before7. The GC retention time and the unique m/z peaks for each amino acid are illustrated in Figure 3.
- Derivatization of amino acids or central metabolites introduces significant amounts of naturally-labeled isotopes, including 13C (1.13%), 18O (0.20%), 29Si (4.70%), and 30Si (3.09%). The measurement noise from natural isotopes in the raw mass isotopomer spectrum can be corrected by using published software5, 8. The final isotopic labeling data are reported as mass fractions, e.g., M0, M1, M2, M3 and M4 (representing fragments containing zero to four 13C labeled carbons).
- Measurement of amino acids can provide isotopic labeling information about eight crucial precursor metabolites: 2-oxo-glutarate, 3-P-glycerate, acetyl-CoA, erythrose-4-P, oxaloacetate, phosphoenolpyruvate, pyruvate, and ribose-5-P. The labeling patterns in these metabolites can be used to identify several central metabolic pathways (Figure 2) 9. The outcome of the labeling experiments can be further confirmed using other biochemistry methods (e.g., RT-PCR).
5. Pathway analysis using labeled amino acid data
By investigating only a few key amino acids produced from well-designed 13C tracer experiments, we may reveal several unique pathways or enzyme activities without performing sophisticated 13C-metabolic flux analysis of entire central metabolism.
- Entner–Doudoroff pathway: [1-13C] glucose can be used as the carbon source. If the pathway is active, serine labeling will be significantly lower than labeling in alanine 10.
- Branched TCA cycle: [1-13C] pyruvate can be used as the carbon source. If the TCA cycle is broken, aspartate can be labeled by two carbons, while glutamate is labeled with only one carbon 11, 12.
- CO2 fixation by Calvin-Benson-Bassham cycle in a mixotrophic metabolism: Non-labeled CO2 and labeled carbon substrates are both used as the carbon sources. If Calvin cycle is functional, serine and histidine labeling will be significantly diluted, comparing to other amino acids. Such method can determine the relative CO2 fixation when organic carbon sources are present in the medium 13.
- Oxidative pentose phosphate pathway: [1-13C] glucose can be used as the carbon source. If the pathway is active, non-labeled alanine will be >50% 12.
- Anaplerotic pathway (e.g., PEP + CO2 à oxaloacetate): 13CO2 and non-labeled carbon substrates (e.g., glycerol or pyruvate) can be used as the carbon source. If the pathway is active, aspartate labeling will be significantly enriched, comparing to alanine and serine 13.
- Re-citrate synthase: [1-13C] pyruvate can be used as the carbon source. If the enzyme is active, glutamate is labeled in β-carboxyl group 3, 4.
- Citramalate pathway: [1-13C] pyruvate, [2-13C] glycerol, or [1-13C] acetate can be used as the carbon source. If the pathway is active, leucine and isoleucine labeling amounts are identical 14.
- Serine-isocitrate lyase cycle: [1-13C] pyruvate or [1-13C] lactate can be used as the carbon source. If the pathway is active, the third position carbon in serine will be labeled 15.
- Utilization of nutrients (i.e., exogenous amino acids): a culture medium with fully labeled carbon substrates and non-labeled amino acids can be used. If the cells selectively utilize these supplemented non-labeled nutrients, we will see significant labeling dilution of these amino acids in the biomass. This method can be used to investigate which nutrient supplements are preferred by the cell 16.
6. Representative Results
Recent bioenergy studies have revived interests in using novel phototrophic microorganisms for bioenergy production and CO2 capture. In the past years, quite a few 13C-metabolism analyses, including advanced 13C-Metabolic Flux Analyses (13C-MFA), have been applied to investigate central metabolisms in phototrophic bacteria, because biochemical knowledge of the central metabolic pathways is not well-founded in these non-model organisms10, 11, 17-20. Here, we present an example of the discovery of an alternate isoleucine pathway in Cyanothece 51142 21. Cyanothece 51142 does not contain the enzyme (EC 4.3.1.19, threonine ammonia-lyase), which catalyzes conversion of threonine to 2-ketobutyrate in the typical isoleucine synthesis pathway. To resolve the isoleucine pathway, we grow Cyanothece 51142 (20 mL) in ASP2 medium 22 with 54 mM glycerol (2-13C, >98%). Cyanothece 51142 utilizes 2nd position labeled glycerol as the main carbon source. We observe that threonine and alanine have one labeled carbon, while isoleucine is labeled with three carbons. Therefore, synthesis in Cyanothece 51142 cannot be derived from the threonine route employed by most organisms (Figure 4). On the other hand, leucine and isoleucine have identical labeling patterns based on fragment (M-15)+ and fragment (M-159)+. For example, the isotopomer data from [M-15]+ (containing unfragmented amino acids) show identical labeling for leucine (M0=0.01, M1=0.03, M2=0.21, M3=0.69) and isoleucine (M0=0.01, M1=0.03, M2=0.24, M3=0.67). Thus leucine and isoleucine must be synthesized from the same precursors (i.e., pyruvate and acetyl-CoA). This observation is consistent with the labeled carbon transition in the citramalate pathway for isoleucine synthesis. To confirm this pathway, we search the Joint Genome Institute database and find the presence of a citramalate synthase CimA (cce_0248) in Cyanothece.
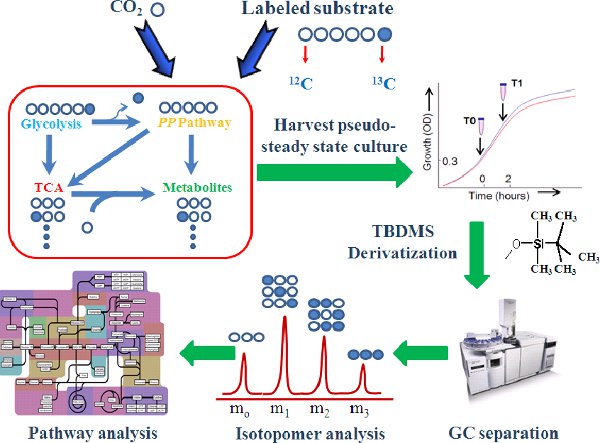
Figure 1.
The 13C-assisted pathway analysis steps.
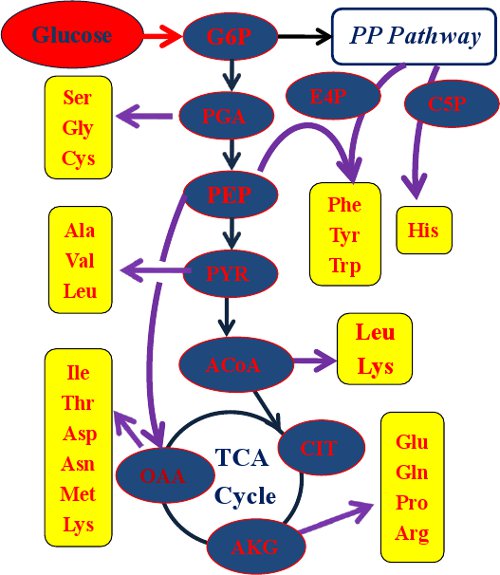
Figure 2.
Amino acids used for acquiring the labeling pattern of their metabolic precursors. ACoA, acetyl-CoA; AKG, α-Ketoglutarate; C5P, ribose 5-phosphate; CIT, citrate; E4P, erythrose 4-phosphate; G6P, glucose 6-phosphate; OAA, oxaloacetate; PEP, phosphoenolpyruvate; PGA, 3-phosphoglycerate; PYR, pyruvate.
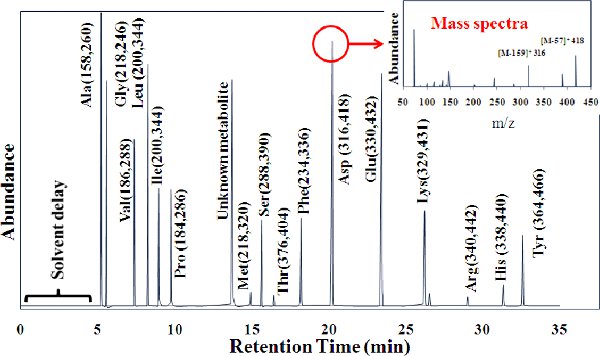
Figure 3. GC peaks for 16 amino acids. TBDMS derivatized amino acids are cracked by MS into two fragments: (M-57)+, containing the entire amino acid, and (M-159)+, which lacks the α carboxyl group of the amino acid. For leucine and isoleucine, the (M-57)+ was overlapped by other mass peaks. We suggest using fragment (M-15)+ to analyze the entire amino acid labeling. The (f302)+ group is detected in most amino acids, which contains only the first (α-carboxyl group) and second carbons in an amino acid backbone. Because this MS peak often has high noise-to-signal ratios, (f302)+ is not recommended for quantitatively analyzing the metabolic fluxes7.
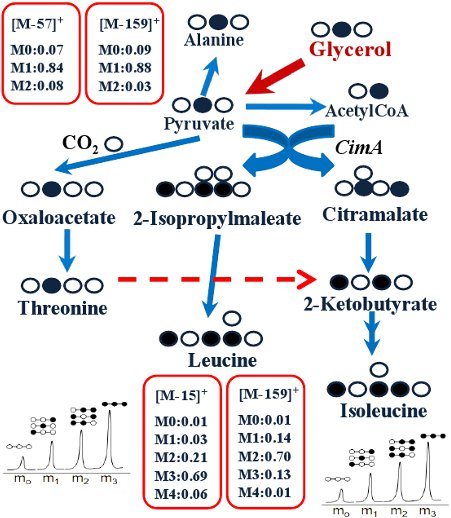
Figure 4.
Labeling transitions in isoleucine pathways in Cyanothece 51142 (modified from our previous paper)21.
