In Figure 1 we show the result of dermal-specific lentiviral shRNA knockdown of Smoothened (Smo), a critical Shh signaling component, in a 3-week old regenerative hair graft. Control grafts using lentiviral vector alone show robust hair growth (Figure 1A). By contrast, dermal-specific knockdown of Smo results in loss of hair growth (Figure 1B). Hematoxylin and eosin stain depicts the stage of hair follicles in control and experimental conditions (Figure 1C, D). The hair growth phenotype from similar treatments can be variable among experiments, including the positive control with lentiviral vector alone infection. Therefore positive control grafts should always be included in every experiment as a reference as 30% of grafts can fail as a result of scarring or incomplete attachment of grafts. In the case of testing an interaction between two factors such as overexpressing cDNA of one factor to rescue shRNA-mediated knockdown of another factor, one should always include knockdown alone grafts to manifest the loss-of-function result in each experiment. This will provide a necessary range to determine how a particular gene perturbs the hair follicle regeneration pathway and how two genes interact.
Removal of the chamber (Figure 2A) must be done with care. The newly formed graft should be slightly opaque with a dull surface (Figure 2B). The graft may stick to the chamber, so gently lifting one side is important to make sure the graft remains attached to the wound bed. The graft is quite stable after three days and should present little difficulty when removing the dressing. Robust hair growth should be observed after three weeks (Figure 2C). Omitting either mDCs or KCs will result in a recovered wound site without any hair (Figure 2D)11. Cell death or cell loss in one of the cell types also results in no hair, in contrast to reduced hair regeneration in dermal-Smo knockdown as shown in Figure 1B.
To ensure a successful assay, it is critical to achieve greater than 90% viral infection of cells with appropriate overexpression or knockdown efficiency before grafting. Due to the time constraints of the grafting procedure, we recommend trouble shooting viral infection efficiencies before starting a grafting experiment, and always save a small portion of cells on day of grafting procedure to verify loss- or gain-of-expression. Lentiviral titer will vary depending on the construct used and should be determined before infection to achieve 90-100% efficiency. We monitor viral infection efficiency with GFP coexpressed from the lentiviral vector. We routinely perform quantitative PCR and/or Western blot to determine the extent of knockdown or overexpression. Utilizing dual-expression vectors with fluorescent tags to label virally infected cells provides a way to report perdurance of signal and number of cells expressing our constructs in mature grafts. We use GFP driven by a CMV promoter from the lentiviral vector pSicoR-CMV-GFP 10 in combination with Smo shRNA. In Figure 3, we show a 3-week old dermal-specific graft where GFP is lentivirally-expressed in dermal papilla of a representative hair follicle.
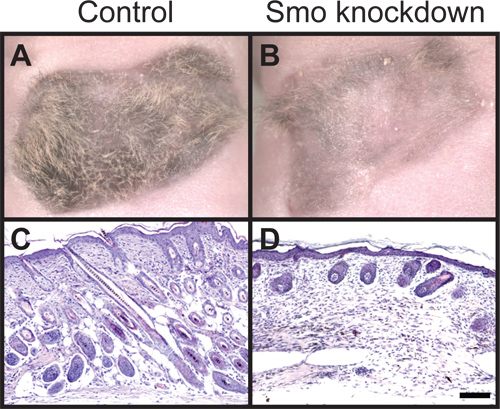
Figure 1. Histological analysis of hair follicle growth. (A) View of control hair regeneration graft with dermal cells infected with empty lentiviral vector and untreated keratinocytes. (B) Smoothened knockdown using lentiviral shRNA in dermal cells combined with untreated keratinocytes results in loss of hair follicles. (C) Hematoxylin and eosin staining shows anagen hair follicles in control grafts extend down into the dermis and (D) Smoothened knockdown hair follicles remain stunted and largely absent. All images are three weeks after grafting. Scale bar denotes 100 μm.
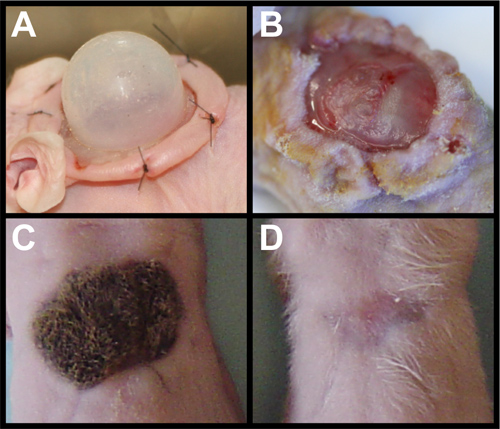
Figure 2. Grafting primary cells. (A) nu/nu mouse with sutured chamber housing primary dermal cells and keratinocytes. (B) Removal of chamber exposes newly formed skin that is dull and opaque in character. (C) Fully grown hairs on graft using wild-type primary dermal cells and keratinocytes at three weeks after grafting. (D) Addition of primary dermal cells without keratinocytes leads to no hair after three weeks. Similar results are seen with addition of primary keratinocytes without dermal cells.
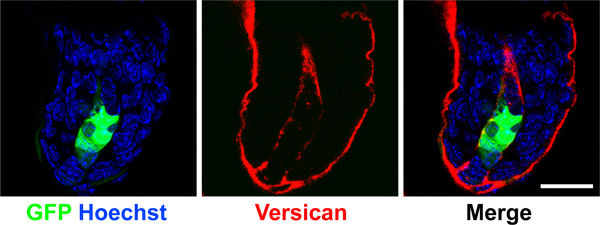
Figure 3. Maintenance of GFP expression in dermal papilla. Confocal images of dermal papilla from a 3-week old regenerated graft. GFP was expressed in the dermal cell population from a lentiviral vector and detected in the dermal papilla (green). Versican (red) demarcates the dermal papilla and nuclei were label with Hoechst (blue). Note GFP is expressed specifically in dermal cells but not in the surrounding keratinocytes. Scale bar denotes 20 μm.
