The 3DNA software tools are routinely used to analyze nucleic acid structures. For example, the identities of base pairs and the rigid-body parameters that characterize the arrangements of bases in double-helical fragments of DNA and RNA structures are automatically computed and stored for each new entry in the Nucleic Acid Database22, a worldwide repository of nucleic acid structural information. The values of the rigid-body parameters determined with Protocol 2 readily reveal distortions in three-dimensional structure, such as the two sites of extreme DNA bending into the major groove, with large positive roll angles (64.95° and 60.93°), found at AT·AT steps 13 and 22 in the crystal complex with the Borrelia burgdorferi Hbb protein8 (Figure 1).
The capability of the software to rebuild structures from these quantities with Protocol 3 makes it possible to determine how individual base and base-pair steps contribute to the overall molecular fold. As illustrated in Figure 2, the global bending of DNA induced by Hbb reflects more than the two extreme roll distortions noted above. That is, the DNA remains highly curved when reconstructed with these base-pair steps straightened, i.e. with null roll angles at the two sites. The same technique has previously revealed the contributions of specific base-pair steps and deformations to the superhelical pitch of the DNA wrapped on the surface of the nucleosome core particle6 and to the width of the minor groove of the DNA bound to the bacterial nucleoid-associated protein Fis23.
The new capability in 3DNA, described in Protocol 4, to examine large numbers of related structures makes it possible to extract both sequence- and time-dependent patterns in the spatial arrangements of simulated DNA and RNA molecules. For example, the (yellow) color-coding of the roll angles between successive base pairs in two large sets of simulated DNA structures11 reveals the preferential bending of these molecules at pyrimidine-purine base-pair steps (Figure 3). The higher values of roll, depicted in red, that persist for short periods at the ends of the DNA are suggestive of localized melting and reannealing of the double-helical structure. The variational patterns of other rigid-body parameters, such as the angles and distances between complementary bases, can help to decipher the precise structural distortions.
The capability of the 3DNA software, presented in Protocol 5, to reorient related molecules in a common reference frame, reveals features of overall structure hidden in many of the files stored in the PDB. For example, the conventional alignment of related structures on the basis of a root-mean-square fit of corresponding atoms produces a series of similar spatial pathways that roughly superimpose upon one another, here the ten NMR-based models of the O3 DNA operator bound to the headpieces of the Lac repressor protein12 (Figure 4 left). The superposition of the same structures on a common coordinate frame on the 5´-terminal base pair of each duplex reveals sizable distortions of global structure, in which the molecules flex in appreciably different directions (Figure 4 right). The structural variability may influence the ease with which the Escherichia coli Lac repressor protein binds O3 and induces a loop between O3 and sequentially distant operators in the lac operon24.
The steps outlined in Protocol 6, for building models of long DNA fragments decorated at arbitrary sites with proteins and other ligands, adds a new perspective on the organization of large macromolecular assemblies. Such models help to understand how the multi-molecular complexes interact during biological processing. As illustrated in Figure 6, the precise placement of an architectural protein like Hbb can have a dramatic effect on the overall folding of DNA. If two copies of the known high-resolution structure8 are separated by 43 base pairs, an 81-base-pair DNA fragment closes into a tight, nearly closed configuration. If the two proteins are separated by an additional five base pairs, the DNA follows an open, meandering pathway. The very different arrangements of the protein-decorated duplex show how the spacing of architectural proteins can affect the cyclization or looping of DNA25, 26.
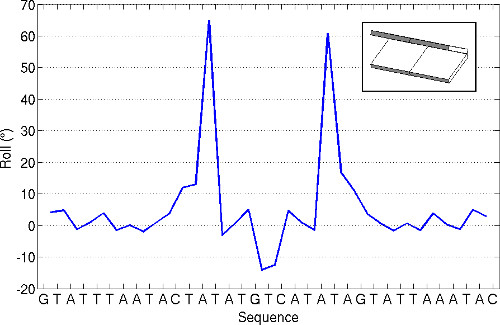
Figure 1. Variation of the roll angle between successive base pairs (see insert for visual depiction) along the DNA chain bound to the Hbb protein from Borrelia burgdorferi8. Values obtained using the ‘analyze’ command of 3DNA and the structural data described in Protocol 2. Note the extreme values of roll at the AT steps that bracket the central third of the structure.
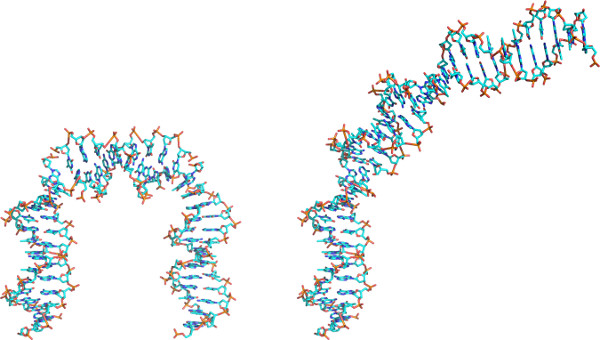
Figure 2. Approximate atomic models of DNA constructed using the ‘rebuild’ function of 3DNA and rendered in PyMOL with color-coded atoms (C-cyan; N-blue; O-red; P-gold). Models based on (left) the rigid-body step parameters of the Hbb protein and (right) a modified set of step parameters, where the two largest values of roll have been set to zero. See Protocol 3 for step-by-step instructions. Note the unfolding of DNA induced by the imposed changes in roll.
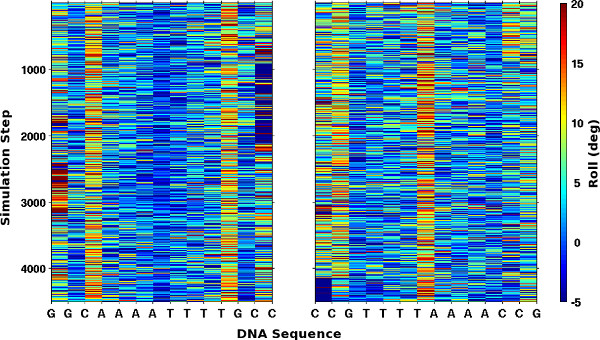
Figure 3. Mosaic images of the roll angles along the DNA in two sets of simulated structures11. Values of roll, extracted using Protocol 4, are color-coded from blue to red over the range [-5°, 20°]. Note the reverse order of bases and large values of roll, highlighted by yellow/red columns, which occur at pyrimidine-purine steps in the two 14 base pair self-complementary sequences.
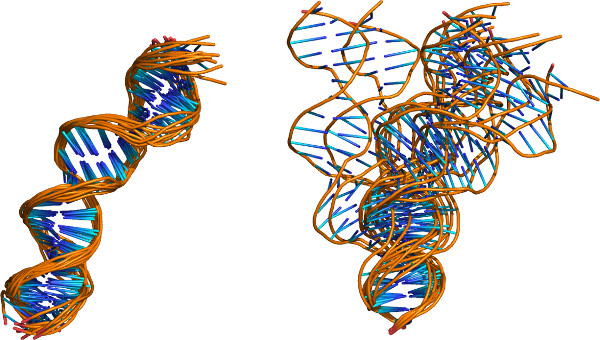
Figure 4. Cartoon images of the DNA models found in the NMR-derived structures of the O3 DNA operator with the Lac repressor protein headpieces12 illustrative of the capabilities of the ‘x3dna_ensemble reorient’ command. Images rendered in PyMOL (backbones show as gold tubes and bases as blue sticks) and aligned using (left) the coordinates in the PDB entry (2kek) and (right) the structural superposition presented in Protocol 5. Note the large differences among the structures when placed in a common reference frame on the 5´-terminal base pair.
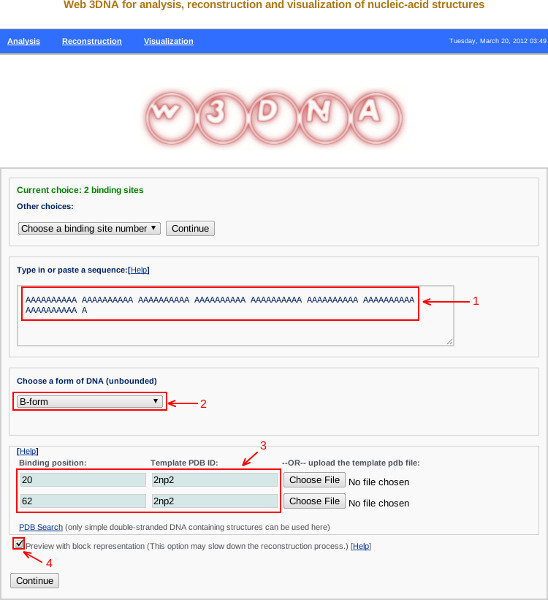
Figure 5. Screen shot from the w3DNA web server illustrating specifications of the DNA sequence (Label 1), the helical form of unbound DNA (Label 2), the positions and identities of proteins (Label 3), and the preview image check box (Label 4) described in Protocol 6. Click here to view larger figure.
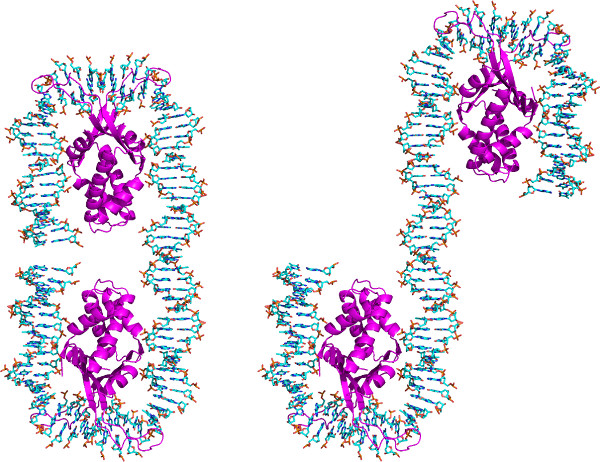
Figure 6. Approximate atomic models of two Hbb proteins8 bound to a long DNA fragment. Structures created with the w3DNA web server as described in Protocol 6 and rendered in PyMOL. The protein chains are show as violet ribbons while the DNA is colored-coded by atom type (C-cyan; N-blue; O-red; P-gold). The central base pair of each protein-binding sites is set at positions (left) 20 and 62 along the 81 base-pair DNA chain and (right) 20 and 67 along the 86 base-pair DNA chain. Note the major change in the folding of the structures associated with the increased (five base-pair) displacement of the two proteins.
