Using a detailed intravital microscopy analysis, heterotypic platelet-neutrophil interactions on the activated endothelium were visualized by infusion of fluorescently-labeled antibodies against a platelet (CD42c) or neutrophil marker (Gr-1) into live mice.
In a model of TNF-α-induced venular inflammation, most rolling neutrophils were stably adhered to the endothelium presumably by interaction of activated β2 integrins with ICAM-1 during the recording period (3-4.5 hr after injection of TNF-α, Figure 2A).8 Rolling and adherent neutrophils were already present before the video capture began due to the TNF-α-induced endothelial cell activation. The number of rolling and adherent neutrophils on the inflamed endothelial cells was 0.25 ± 0.05 cells per minute and 18.5 ± 0.7 cells per 5 min, respectively (Figures 2B and 2C). Most neutrophils were adhered to the activated endothelium for the duration of video capture (1-1.5 hr), and there was only a minimal decrease in adherent cells along with a minimal increase in rolling cells (data not shown). We found that most platelets adhere to adherent and crawling neutrophils rather than the inflamed vessel wall (Figure 2A, Video 1). Platelet thrombi accumulated and embolized repeatedly for the duration of the video capture. The integrated fluorescence signal associated with adherent platelets was shown in Figure 2D.
Utilizing the laser-induced arteriolar thrombosis model, we were able to examine and characterize the heterotypic interaction of platelets and neutrophils at the site of arteriolar wall injury. In this model, the thrombus size peaked around 100 sec after laser injury, followed by a series of rapid small embolization for the next 2-3 min (data not shown).9,10 Five minutes after laser injury, the size of platelet thrombus remained relatively constant during imaging (5-25 min after laser injury, Figures 3A-3C) and neutrophils rolled on and adhered to the platelet thrombus (Figures 3A-3B, Video 2). The fluorescence signal from the circulating platelets was negligible in comparison with that from the platelet thrombus. The number of rolling and adherent neutrophils was 21.5 ± 3.0 and 1.6 ± 0.4 cells over 20 min, respectively (Figures 3D-3E). Initial rapid rolling of neutrophils occurred on the endothelial cells. Once neutrophils contacted the platelet thrombus, the rolling velocity of neutrophils on a platelet thrombus was changed with a range of 8.2 ± 1.1 μm per second (Figure 3F), which is mediated by interaction of P-selectin and PSGL-1.11
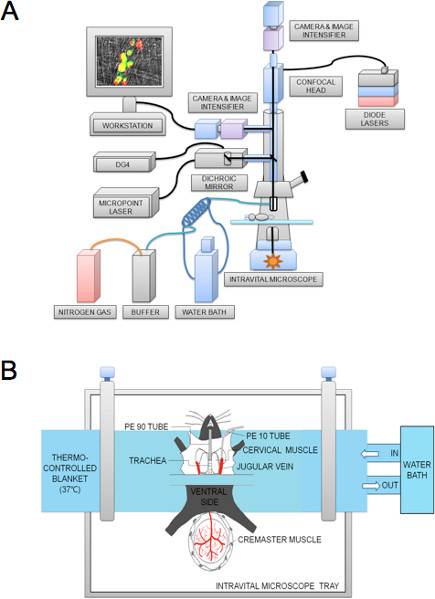
Figure 1. Schematic of the intravital microscope system (A) and preparation of the cremaster muscle microvessel (B).
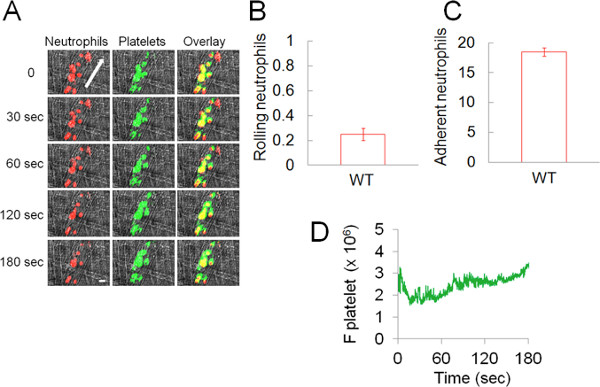
Figure 2. Heterotypic interactions of platelets and neutrophils during the TNF-α-induced venular inflammation in live mice. Platelets and neutrophils were detected by Dylight 488-conjugated anti-mouse CD42c and Alexa Fluor 647-conjugated anti-mouse Gr-1 antibodies, respectively. (A) Representative binarized images of the appearance of fluorescence signals associated with neutrophils (red) and platelets (green) over 180 sec. Arrow shows direction of blood flow. Bar = 10 μm. (B-C) The number of rolling (cells/minute) and adherent neutrophils (cells/5 min) on inflamed endothelial cells is shown. Data represent the mean ± SEM of the 30 different venules in 4 wild-type mice. (D) Median integrated fluorescence signal of platelets (F platelet) is plotted as a function of time. No signal was detected with Dylight 488-conjugated control rat IgG (data not shown).
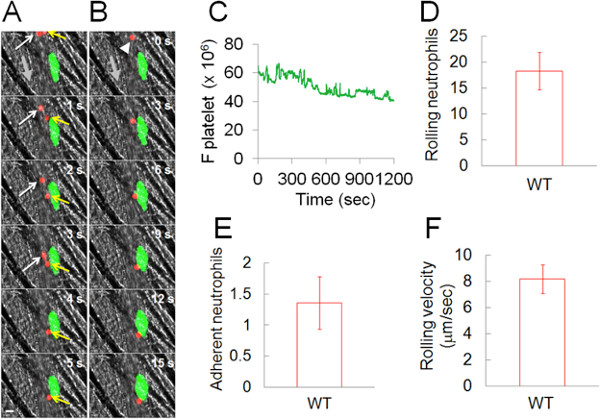
Figure 3. Heterotypic interaction of neutrophils with a platelet thrombus at the site of laser-induced arteriolar injury in live mice. Platelets and neutrophils were detected as described in Figure 2. (A) A single neutrophil (red, a yellow arrow) rolls over the platelet thrombus (green) while a second neutrophil (red, a white arrow) rapidly rolls over arteriolar endothelial cells, shown over 5 sec. (B) A single neutrophil (red) rolling over and adhering to a platelet thrombus (green) shown over 15 sec. The arrowhead shows rolling and adherent neutrophils. Bar = 10 μm. The thick, grey arrow shows the direction of blood flow. (C) Median integrated fluorescence signal of platelets (F platelet) is plotted as a function of time. (D-E) The number of rolling and adherent neutrophils on the platelet thrombus is shown (cells/20 min). (F) The rolling velocity of neutrophils over the platelet thrombus. Data represent the mean ± SEM of the 14 thrombi in 5 wild-type mice.
