Investigating the role of enzymes of the Doa10 pathway in the degradation of a reporter substrate
To test the validity of the GiLS method it was compared to a traditional degradation assay. This experiment assesses the relative contribution of components of the ER-membrane localized Doa10 E3-ligase complex20,21 to the degradation of the protein quality control reporter substrate Deg1-VU (Figure 3A). The Deg1-VU plasmid was integrated into wild type cells or into cells lacking the gene for the E2 conjugating enzyme, UBC7 (ubc7) and protein stability was subsequently determined by a CHX chase assay6. Anti-FLAG immunoblotting revealed a stable Deg1-VU protein band in ubc7 cells that was absent in wild type cells (Figure 3B). The absence of Deg1-VU in wild type cells is attributed to its continuous degradation that was abolished in ubc7 cells, indicating that Deg1-VU stability is strongly dependent on Ubc7.
Obviously, the degradation of Deg1-VU is rapid. However, the relative rate of degradation cannot be determined by a CHX assay since the protein is undetectable in wild type cells from the outset. Consequently, the newly-developed GiLS assay method was employed, in order to compare the relative differences in the kinetics of protein degradation between wild-type and various mutant strains (Figure 3C). Initially, wild type, ubc6, ubc7 or doa10 cells expressing Deg1-VU were grown on SD complete medium. To measure GiLS, cells were washed and incubated in SD-URA. MDT calculation was executed as exemplified in the protocol. Figures 3C, 3D show similar growth curves and calculated MDT (~2.5 hr) for all strains grown in SD complete, excluding the possibility that the deletion of any of the examined genes affects cell growth. In contrast, incubation in SD-URA resulted in poor growth of wild-type cells (MDT=6.34), whereas only a minor growth defect was observed in the mutant strains (MDT ~3 hr).
Examining the effect of different mutants of the E2 conjugating enzyme Ubc7 on the degradation of Deg1-VU
The fact that Ubc7 is absolutely required for Deg1-VU degradation enables accurate assessment of even partial effects of various E2 mutants, since the experimental range of the system is very wide. Accordingly, plasmids containing wild type or mutant UBC7 were integrated into Ubc7 cells expressing Deg1-VU and their contribution to degradation was determined by GiLS. As expected, fast growth kinetics were observed in cells expressing Ubc7 with the active-site mutants C89S and N81A22 (Figure 4). In addition, two mutants predicted to indirectly hinder Deg1-VU degradation (V25G and H94K) were tested. These mutations also enhanced cell growth, compared to wild-type Ubc7, albeit to a lesser degree than the active-site mutants (average MDTs of 3.4 hr for V25G and H94K compared to average MDTs of 2.7 hr for C89S and N81K) (Figure 4). Thus, the GiLS method can be used for accurate measurement of the relative contribution of various degradation factors to the stability of a reporter substrate.
Isolation and evaluation of novel degrons
The GiLS system was also employed in a high throughput screen format to identify novel degrons and quantify their relative potency, i.e., the degree by which they induce degradation (Figure 5). To optimize the identification of bona fide degrons, an additional selection step, which is growth in the presence of Fluoroorotic Acid (5-FOA), was added. Ura3 efficiently converts 5-FOA into a toxic compound (5-fluorouracil) that can serve as a positive selection for the isolation of yeast strains where Ura3 is destabilized16,23. To identify novel degrons, a library of reporter plasmids was generated by fusing random fragments derived from a yeast cDNA library to Ura3-GFP plasmid24 (Figure 5A). The GFP moiety was added in order to enable a secondary screen and provide information about the localization of the fusion degron (see discussion). The library was transformed into yeast, then selected in the presence of 5-FOA (Figure 5B) and positive clones were isolated and re-seeded on agar plates in a 96-well plate format. Each colony was transferred into 96-well plate, incubated O/N, and diluted in a new 96-well plate as described in section 2.2. It is expected that growth of cells in SD-URA medium will correlate with the degree of Ura3 expression, which is the outcome of the degron's potency to induce degradation. This was subsequently confirmed by applying GiLS (Figure 5C). We confirmed that all randomly-picked colonies that were pre-selected with 5-FOA showed similar MDT values, on SD-LEU medium (Figure 5C, empty bars). In contrast, all clones showed variable growth rates on SD-URA, which were significantly slower than cells expressing control Ura3-GFP (Data not shown). As indicated, there is an inverse relationship between growth rate on SD-URA and MDT. Thus, the higher the MDT, the more potent is the degron. Indeed, all MDTs derived from positively selected clones were higher than that of the control (above the red line) (Figure 5C, filled bars).
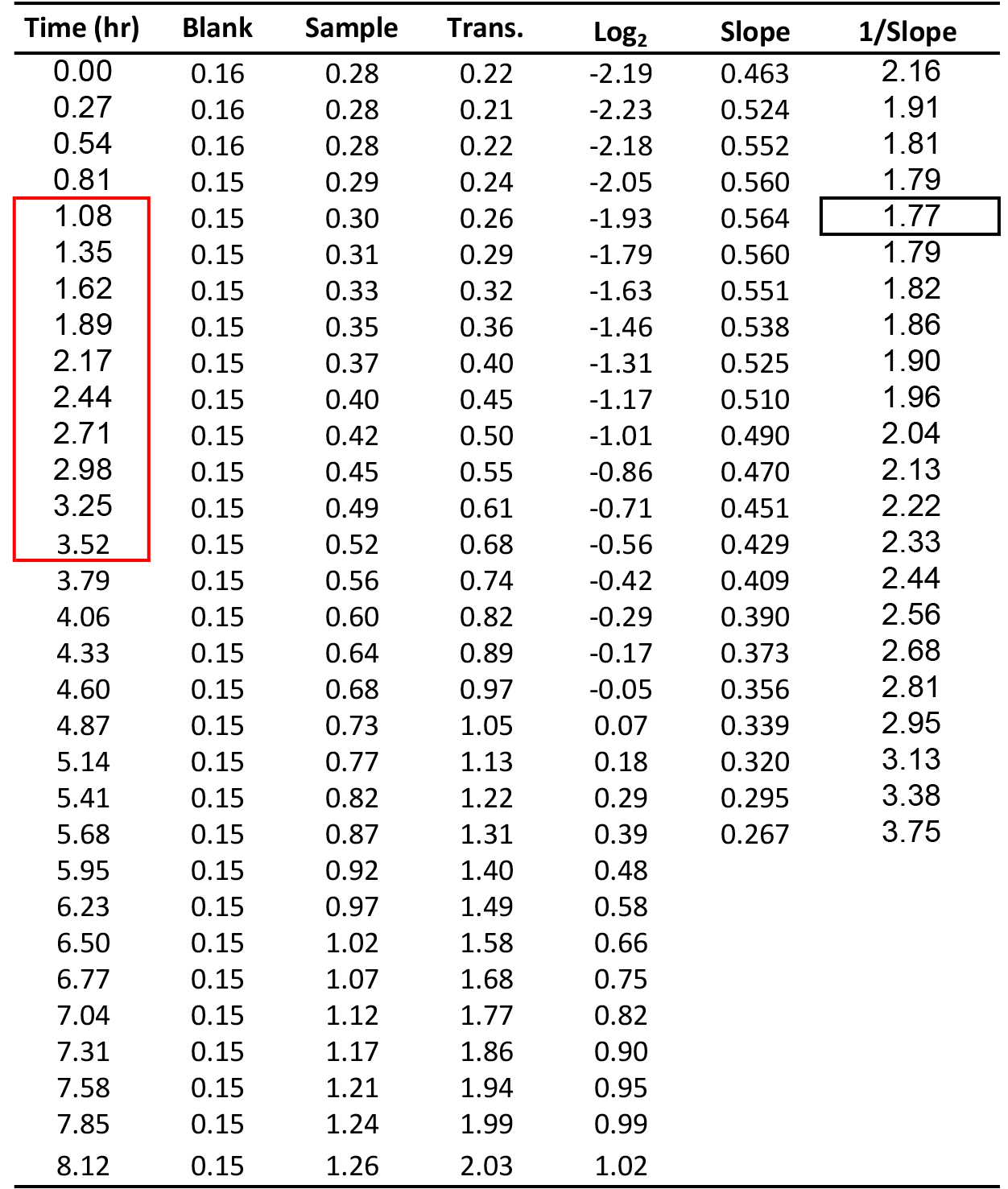
Table 1. Illustration of the calculation of yeast MDT. Time units are hours (hr). Blank and sample are from OD600 reads. Transformation (Trans.) is the OD600 value after blank subtraction and path-length correction. Log2 is calculated from the transformed data. Slope is the Log2 (OD600) values within consecutive of intervals of 10 time points divided by time. 1/Slope is the time required for the cell population to duplicate itself. Black rectangle marks the MDT value. Red rectangle marks the time interval from which the MDT was calculated.
| Yeast | Genotype | Source |
| TRy467 | α, his3∆1, lys2∆0, ura3∆0, leu2∆0 | Euroscarf |
| TRy508 | α, his3-∆200::pRS303/Ubc7prom-Ubc7-3HA::HIS, leu2-3,112, ura3-5, lys2-801::Met25-Deg1-Flag-Vma12-Ura3::LYS, trp1-1, ubc7∆::LEU2 | This study |
| TRy528 | α, his3-∆200::pRS303/Ubc7prom-Ubc7[N81A]-3HA::HIS, leu2-3,112, ura3-5, lys2-801::Met25-Deg1-Flag-Vma12-Ura3::LYS, trp1-1, ubc7∆::LEU2 | This study |
| TRy530 | α, his3-∆200::pRS303/Ubc7prom-Ubc7[C89S]-3HA::HIS, leu2-3,112, ura3-5, lys2-801::Met25-Deg1-Flag-Vma12-Ura3::LYS, trp1-1, ubc7∆::LEU2 | This study |
| TRy532 | α, his3-∆200::pRS303/Ubc7prom-Ubc7[H94K]-3HA::HIS, leu2-3,112, ura3-5, lys2-801::Met25-Deg1-Flag-Vma12-Ura3::LYS, trp1-1, ubc7∆::LEU2 | This study |
| TRy556 | α, his3-∆200::pRS303::HIS, leu2-3,112, ura3-5, lys2-801::Met25-Deg1-Flag-Vma12-Ura3::LYS, trp1-1, ubc7∆::LEU2 | This study |
| TRy563 | α, his3-∆200::pRS303/Ubc7prom-Ubc7-3HA::HIS, leu2-3,112, ura3-5, lys2-801::Met25-Deg1-Flag-Vma12-Ura3::LYS, trp1-1, ubc7∆::LEU2, ubc6∆::KanMX | This study |
| TRy633 | α, his3-∆200::pRS303/Ubc7prom-Ubc7[V25A]-3HA::HIS, leu2-3,112, ura3-5, lys2-801::Met25-Deg1-Flag-Vma12-Ura3::LYS, trp1-1, ubc7∆::LEU2 | This study |
| TRy786 | α, his3-∆200::pRS303/Ubc7prom-Ubc7-3HA::HIS, leu2-3,112, ura3-5, lys2-801::Met25-Deg1-Flag-Vma12-Ura3::LYS, trp1-1, ubc7::LEU, doa10∆::HIS3 | This study |
Table 2. List of yeast strains used in this study.
| Plasmids used in this study | ||
| Plasmid | Relevant markers | Source |
| pTR717 | YDpK-MET25p-Deg1-Flag-Vma12-Ura3 (integrative plasmid containing LYS1) | This study |
| pTR1412 | pRS315-CUP1p-URA3-HA-GFP (LEU) | This study |
| Additional suitable reporter plasmids | ||
| Plasmid | Relevant markers | Source |
| pOC9-CL1 | pOC9-CUP1p-HA-URA3 (TRP1) | (17) |
| URA3-2(GFP) | pPS414-TRP1p-MYC-ura3-2-GFP | Lewis and Pelham |
Table 3. List of plasmids used in this study. Reference: Lewis, M. J. & Pelham, H. R. Inefficient quality control of thermosensitive proteins on the plasma membrane. PLoS One 4, e5038, doi:10.1371/journal.pone.0005038 (2009).
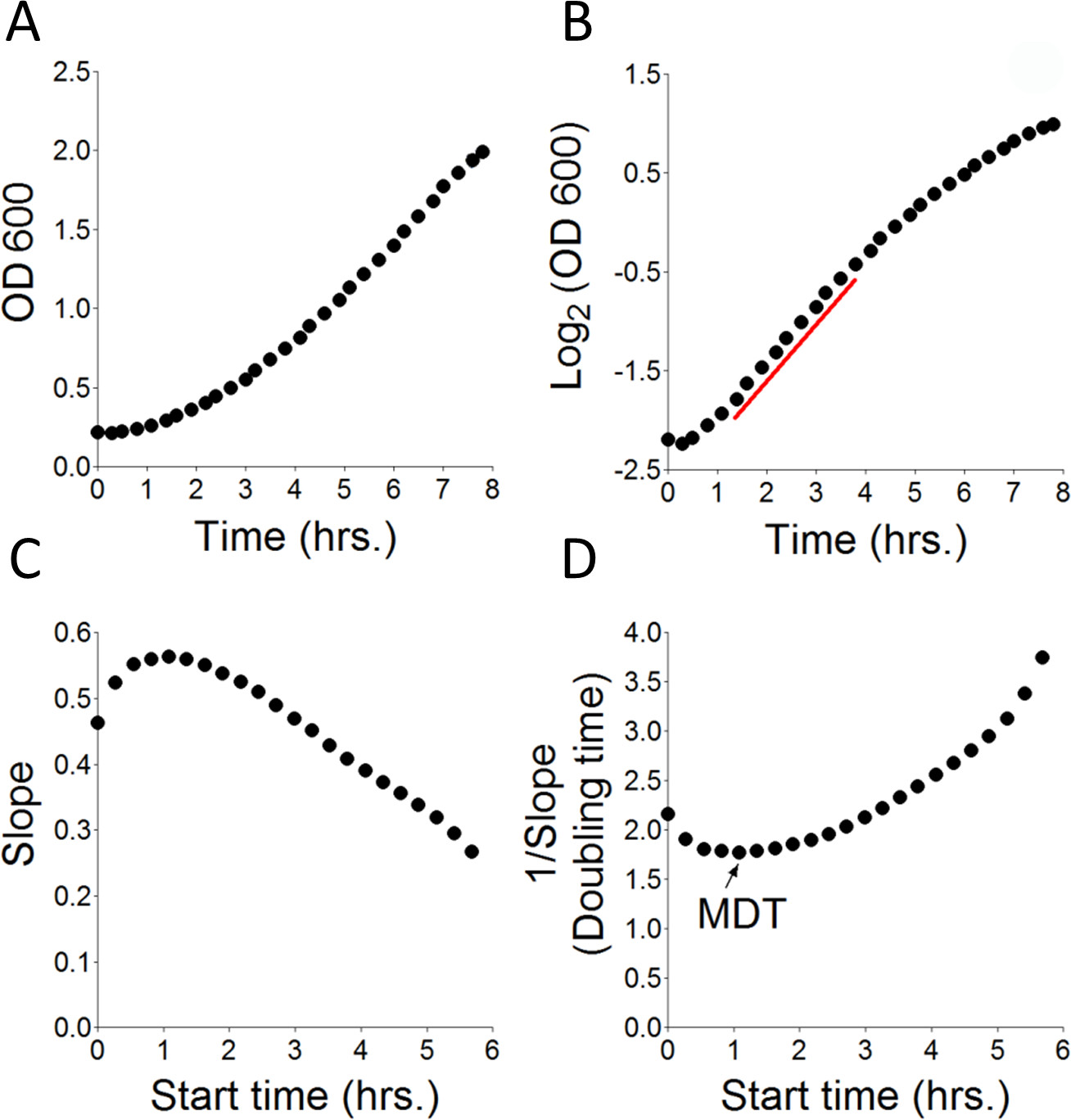
Figure 1. Principles of calculation of MDT. Try467 yeast cells at OD600 0.22, were incubated in SD complete media for the indicated time period. OD600 measurements were taken every ~15 min and the data was collected in a spreadsheet. (A) Transformed OD600 values from Table 1 plotted against time. (B) Log2 of OD600 plotted against time. Red line: Marks the time interval from which the MDT was calculated (D). (C) Yeast growth rate (slope) was calculated by measuring changes in OD600 of consecutive 10 time points (assuming linearity) over time. Start time: The time from which calculation was initiated. (D) The time required for yeast to double is the inverse of the slope from (C) (1/Slope). MDT: The calculated Minimal Doubling time (in hr).
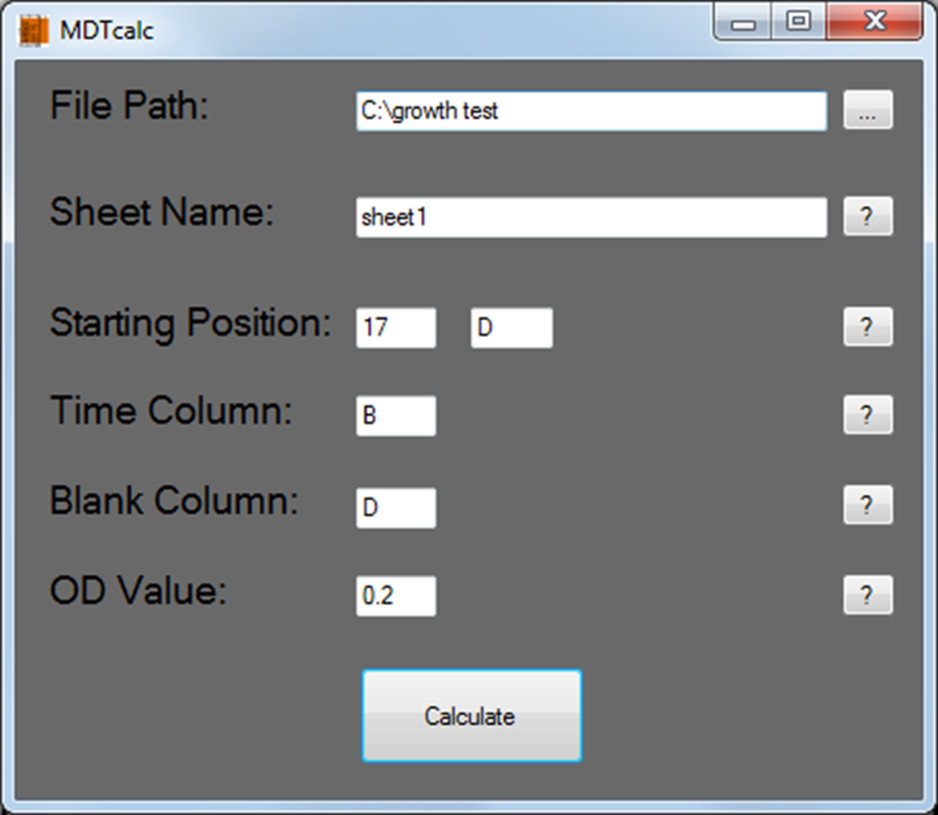
Figure 2. MDTcalc start screen: Protocol section 5.1. Typical input values to be entered are shown.
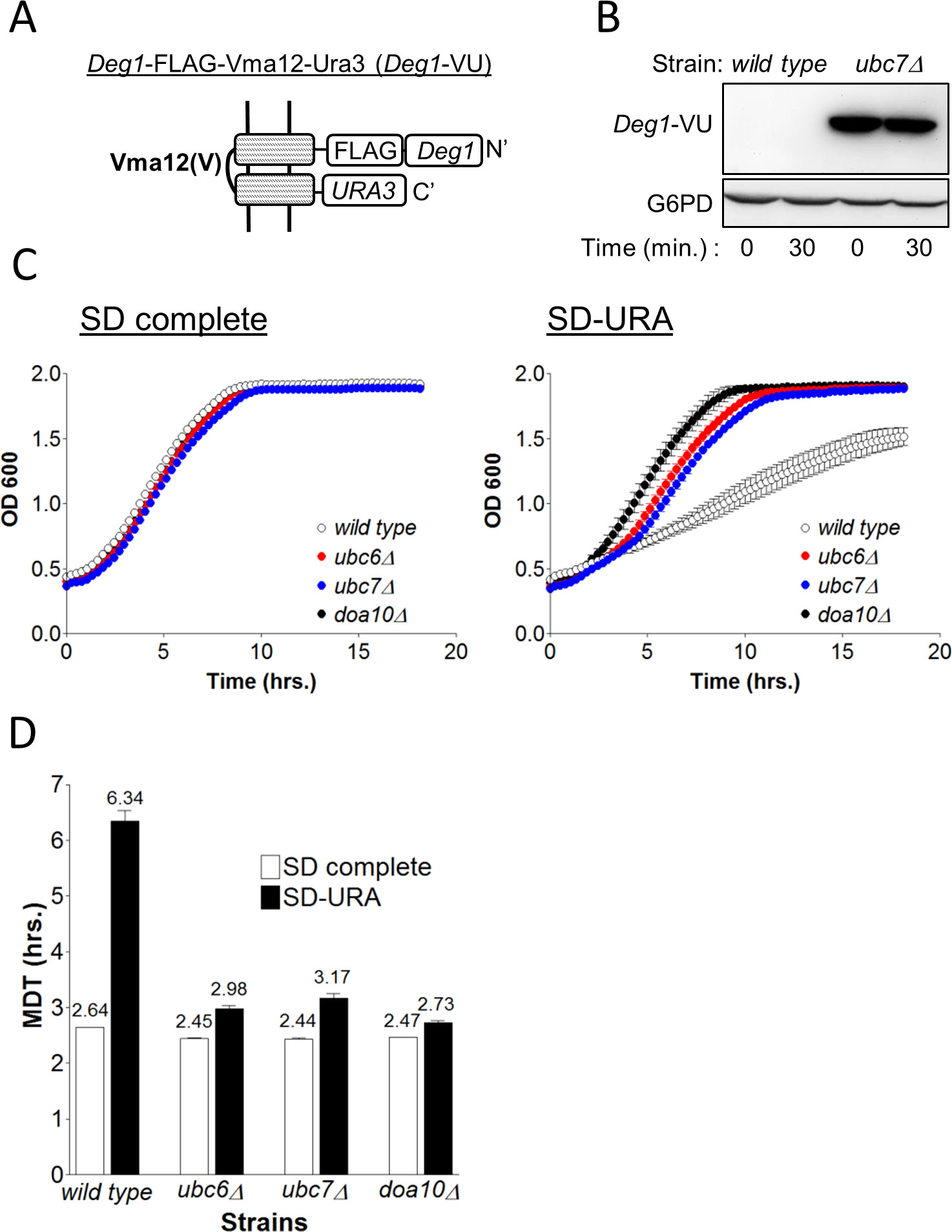
Figure 3. Measuring the degradation of Deg1-VU. (A) Schematic presentation of the different protein elements of Deg1-VU and its organization at the ER membrane.(B) Determination of the degradation of Deg1-VU by the CHX assay in wild type and ubc7 cells. Cells collected at time zero or after 30 min with CHX were lysed and proteins were separated by 5-15% SDS PAGE. Deg1-VU was visualized by immunoblotting using anti FLAG antibodies. G6PD staining served as a loading control. (C) The indicated strains, expressing Deg1-VU were incubated in SD-complete or SD-URA media for 20 hr and OD600 was measured every 15 min. OD600 are the transformed values. (D) The MDT for each strain was calculated using MDTcalc.
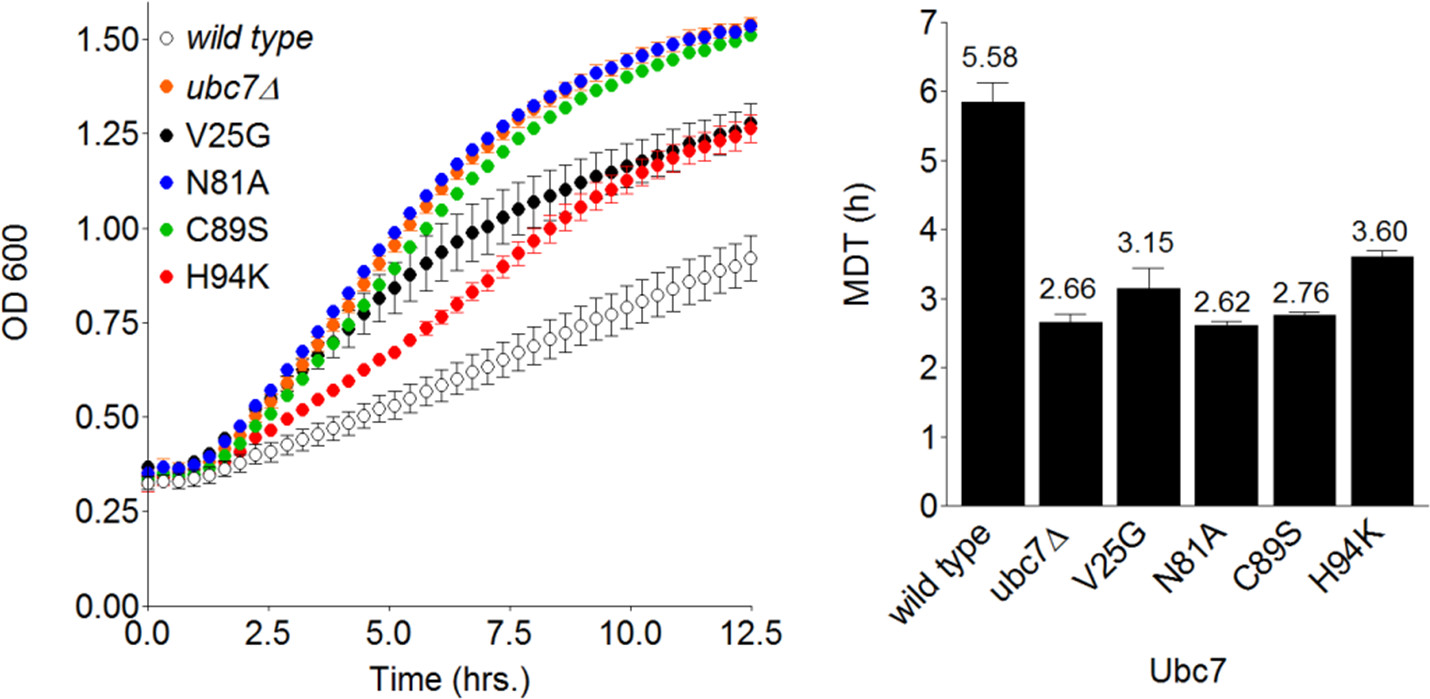
Figure 4. Measuring relative degradation rates of Deg1-VU in Ubc7 mutant strains. Left: Plot of the transformed OD600 values, measured during the replication of wild-type, ubc7Δ and the indicated Ubc7 mutants, versus time. Cells expressing Deg1-VU were grown on SD-URA medium and OD600 was measured every ~15 min. Right: The MDT for each strain was calculated using MDTcalc.
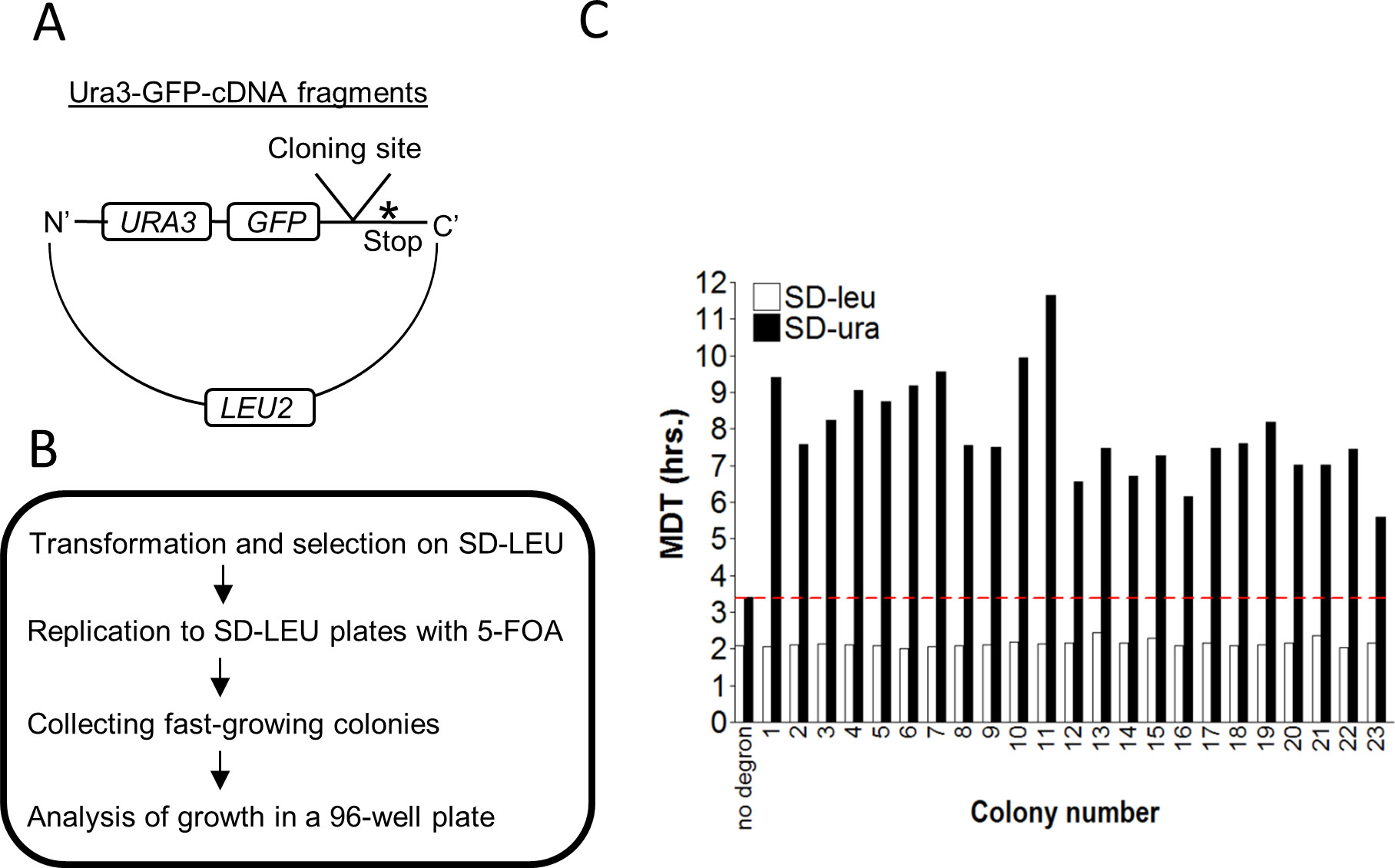
Figure 5. A high throughput assay for identification and isolation of novel degrons. (A) Schematic presentation of the features of the Ura3-GFP-cDNA reporter. (B) A flow chart describing the experimental steps for the isolation of yeast strains carrying a degron. The plasmid library described in A was transformed into Try467 cells, followed by selection on SD-LEU plates. Colonies were replicated to SD-LEU plates containing 5-FOA and fast-growing colonies were collected and organized on plates in an ordered array. (C) Colonies expressing Ura3-GFP-cDNA that grew on plates containing 5-FOA were incubated in either SD-LEU or SD-URA media for 20 hr. OD600 was measured every ~15 min and the minimal yeast doubling time (MDT) was calculated using the MDTcalc software. No degron: a plasmid expressing Ura3-GFP without a degron, serves as a positive control for growth. Red line: threshold determined by the MDT value of the control.
