Viral vectors encoding for fluorescent proteins can be used in addition to, or instead of, fluorescent dye. Viral transduction should be performed in advance of the motility assay. Both adherent and non-adherent cell types can be differentially-labeled. The protocol for transduction will depend on the type of vector employed. Here, we transduced the alloCTL in Figures 3, 5 and 6 with RRV-EMD at least two days in advance of the assay using the protocol described12. We also used a lentiviral vector, CMV-Strawberry-IRES-FLUC2, to transduce the tumor cells to express the mStrawberry red fluorescent protein (Figure 4). This transduction was accomplished by adding the vector to the flask of cells for 48 hr, at which point, fresh medium was added. After allowing the cells to recover for two days, limiting dilution was performed to generate a population of 100% transduced cells.
Human alloCTL were made with inactivated stimulator human 13-06-MG malignant glioma cells by MLTR. The alloCTL then were transduced with RRV coding for EMD (RRV-EMD) and later seeded into the center of established monolayers of U-87MG glioma cells with partial MHC-match to 13-06-MG (Figures 3 and 5). Fluorescence microscopy shows cells, initially seeded at the center of the well, migrate away from the center over time. The formation of alloCTL aggregates after 4 hr (Figure 3, white arrows) is likely reflective of autocrine growth patterns exhibited by activated, IL-2-producing T cell populations13.
In another experiment, murine effector alloCTL were made by one-way MLR using haplotype H-2d Balb/c responder splenocytes and haplotype H-2b/k stimulator splenocytes from B6C3F1 mice, the strain to which the TU-2449 glioma is syngeneic. Immediately before seeding through the channel of the manifold onto the tumor cell monolayer, the alloCTL were stained with eFluor 670. The alloCTL (purple cells) were seeded into the center of established monolayers of TU-2449 glioma cells (red) that were 100% transduced with the CMV-Straw-IRES-FLUC2 viral vector, and plated on wells the day before assay. Figure 4A and B show microscopic images taken of the same quadrant of the same well at 1 and 48 hr. Figure 4C and D were generated using ImageJ to convert digitized purple fluorescence intensities into quantitative surface plots that are displayed in three-dimensional format.
Finally, human alloCTL were made with inactivated stimulator human brain-tropic cells derived from the metastatic breast tumor cell line MDA-MB-231BR by MLTR. Figure 6 shows light and fluorescence microscopy images over time of non-adherent CMPTX-labeled alloCTL, partially transduced with RRV-EMD, migrating on ECM proteins extracted from those tumor cells. In (A) the bright field photomicrograph shows focal placement of alloCTL immediately after sedimentation. In (B and F) bright field photomicrographs show, at 4 and 8 hr, respectively, the alloCTL radially migrating on the ECM. The alloCTL density decreases with distance from the center of the well (center of well is top left corner of field in B-I). Panels (C and G) show the entire alloCTL preparation stained with CMTPX; (D and H) show partial transduction of the alloCTL with RRV-EMD as indicated by small numbers of EMD+ T cells, and (E and I) show the merged images of the RRV-EMD transduced alloCTL migrating on the ECM along with the untransduced alloCTL.

Figure 1. Cell sedimentation process. (A) Wells being prepared for sedimentation, (B) a cell sedimentation manifold placed on a slide, (C) non-adherent effector lymphocytes being loaded into the manifold, (D) manual removal of the cell sedimentation manifold, (E) a humidity chamber, (F) slides being placed in humidified incubator. This figure is used with copyright permission from Creative Scientific (http://www.creative-sci.com/). Click here for a larger version of this figure.
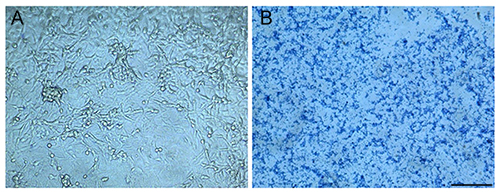
Figure 2. Tumor cell monolayers and ECM plated on CSM slide wells. Photomicrographs of slide wells prepared with (A) confluent U-87MG glioma cell monolayers, or (B) ECM proteins extracted from confluent U-87MG stained with Coomassie blue dye. Bar = 200 μm. Click here for a larger version of this figure.
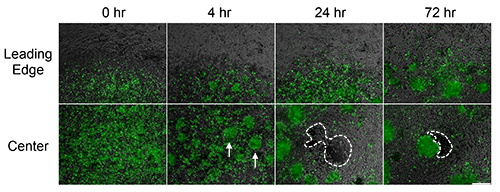
Figure 3. Migration and cytotoxicity of RRV-EMD transduced lymphocytes at the center and leading edge of cell deposit over time. Fluorescent photomicrographs taken at various times at the leading edge and at the center of alloCTL-RRV-EMD following sedimentation on a monolayer of adherent U-87MG glioma cells. Effector alloCTL seeded as single cells, form spherical clusters within 4 hr (white arrows) of placement. Single fluorescently-labeled CTL have migrated from the center of the well as time progresses. After 24 hr, empty cell patches in the adherent monolayer are visible presumably due to alloCTL cytolysis of the tumor target cells (see area within dashes). Bar = 200 μm. Click here for a larger version of this figure.
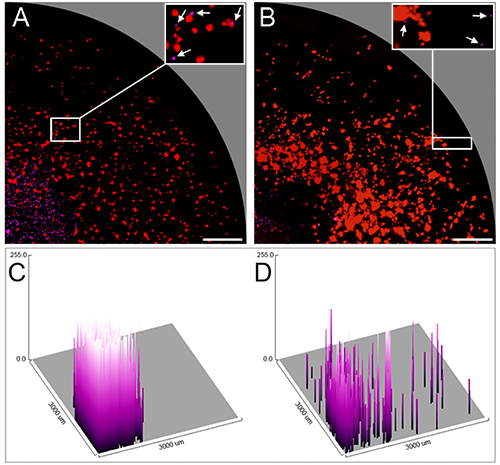
Figure 4. Radial migration of murine alloCTL stained with eFluor 670 on mStrawberry-labeled glioma cells shown at 1 and 48 hr. Fluorescent photomicrographs of one quadrant of the same well at (A) 1 hr and (B) 48 hr after sedimentation of fluorescently-labeled non-adherent alloCTL onto a monolayer of TU-2449 glioma cells. The low power photomicrographs reflect the area contained within one quadrant of the CSM well (radius 3 mm). At high power (A and B, insets) alloCTL (white arrows) are shown in proximity to or associated with individual tumor cells; one has a disintegrated appearance with fragmented nuclei and is apoptotic. (C and D) respective surface intensity fluorescence maps obtained using ImageJ software showing pixel values of the digitized purple fluorescent images in three-dimensional graphic form and placed on a grid representing the well radius of 3 mm. Bar = 500 μm. Click here for a larger version of this figure.
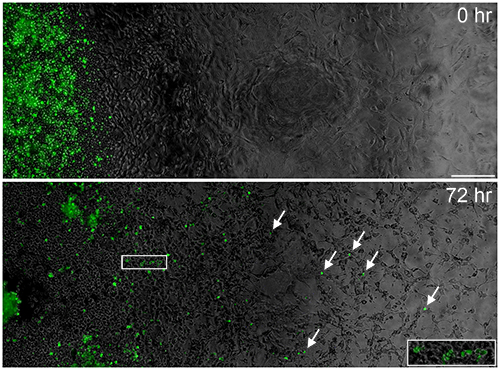
Figure 5. Migration of alloCTL and horizontal spread of RRV-EMD from transduced alloCTL to tumor cells. Fluorescent photomicrographs serially stitched together to cover the radius of the well using image editing software. Migration of RRV-EMD transduced alloCTL (white arrows) is shown on a monolayer of U-87MG tumor cells at 0 and 72 hr following sedimentation. RRV-EMD transduction of tumor cells in the monolayer is also apparent at 72 hr following addition of the EMD-transduced alloCTL, indicating that horizontal transmission of infective RRV from lymphocytes to tumor cells has occurred (white box, and inset). Bar = 200 μm. Click here for a larger version of this figure.
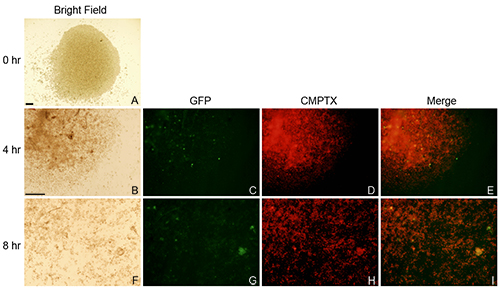
Figure 6. Radial migration of alloCTL transduced with RRV-EMD and stained with CellTracker Red CMTPX. Cells were sedimented onto ECM extracts derived from MDA-MB-231BR cells. Migration of alloCTL was imaged by bright field light microscopy at 0, 4 and 8 hr (A, B, F column 1, respectively). Fluorescent images of the CMPTX (red) labeled alloCTL (C, G, column 2), the RRV-EMD (green) transduced alloCTL (D, H, column 3), and the merged images (E, I, column 4) are shown at 4 and 8 hr. Bars = 200 μm; Bar shown in B applicable to images C-I. Click here for a larger version of this figure.
