In the following section we will present a complete documentation of a GzmA preparation to illuminate the method. We also successfully purified GzmB and GzmM, producing similar results in regard to purification efficiency and activity. However, from those later preparations we will only show a few selected pieces of data. GzmB preparations from 293T cells following the current protocol were used in several published studies, highlighting their activity in various biological assay systems9,29,31-34.
A typical GzmA purification is shown in Figure 2A. Efficient transfection and expression after 72 hr was indicated by a detectable Gzm band on a Coomassie-stained SDS-PAGE gel without further concentration of the culture supernatant (~37 kDa for GzmB and GzmM under reducing conditions; ~60/34 kDa for GzmA under non-reducing/reducing conditions). After the IMAC purification the samples were treated with enterokinase (EK). A visible shift of the EK treated sample (post-EK) compared to the pre-EK control was observed. The final purification on the S-column resulted in highly pure Gzm preparations (GzmA in Figure 2A; GzmB and GzmM in Figure 2B) with a single band appearance on Coomassie-stained protein gels. The pooled fractions were concentrated to a final concentration of ~100 μM. Typically, we obtained yields of about 0.5 to 1 mg Gzms per 100 ml culture supernatant. To demonstrate efficient glycosylation, a few initial preparations were treated with Endoglycosidase (Endo) H, which removes high mannose oligosaccharides from N-linked glycoproteins. Native GzmA has a N-linked glycosylation site at Asn-142, to which a high mannose oligosaccharide is bound35. EndoH treatment induced an obvious mobility shift in the Gzm A, as well as GzmB, preparations from HEK293T cells but not in bacterially generated GzmA as analyzed by SDS-PAGE and Coomassie staining (Figure 2C).
Routinely, every Gzm batch was tested for enzymatic activity in colorimetric assays. This rapid and reliable test demonstrates proteolytic activity of the Gzms by the cleavage of small synthetic peptides that is indicated spectrophotometrically by a color change of the assay buffer. Due to different preferences concerning the amino acid residues (P1 position) after which these proteases cleave, the choice of a specific peptide varies among the Gzms. GzmA has trypsin-like activity, cleaving after the basic residues arginine (Arg, R) and lysine (Lys, K). GzmB cleaves preferentially after aspartic acid residues (Asp, D), and GzmM cleaves after leucine (Leu, L) and methionine (Met, M)1. Our preferential peptide choice is BLT-S-Bzl to measure GzmA activity, AAD-S-Bzl for GzmB and AAPL-pNA for GzmM.
The major difference between the thiobenzylester substrates (AAD and BLT) and the p-nitroanilide substrate (AAPL) is the specific chemistry, in that Gzm mediates cleavage of thiobenzylester substrates releases benzyl-mercaptan, which only triggers a chromogenic reaction in its downstream reaction with the chromophore DTNB. Therefore, in the reaction buffers of thiobenzylester substrates DTNB has to be present, while the Gzm-mediated release of p-nitroanilide is sufficient for the colorimetric detection. The detailed method of the colorimetric assays was recently published36. Figure 3A shows the representative BLT and AAPL esterase activity in the elution fractions from the S-column of a GzmA and GzmM preparation, respectively. Colorimetric assay was also used to compare the activity of recombinant to native GzmB preparations. As shown in Figure 3B, recombinant 293T GzmB cleaved the chromogenic substrate with similar efficiency compared to native GzmB purified from a human NK cell line, as recently described12.
To more specifically test Gzm activity, Gzm cleavage assays using known protein substrates are indicated. These cleavage assays can be performed with purified recombinant or native protein (if available), with cell lysates or most physiological with intact cells. If a protein substrate is available, proteolytic activity can be analyzed in simple co-incubation experiments with the Gzm preparations as this is demonstrated with the known bacterial GzmA and GzmB substrates nuoCD and nuoF9, as well as with the novel human mitochondrial GzmM substrate NDUFAF3 (Figure 4A).
Cell lysates represent another obvious source of multiple Gzm substrates. Commercial antibodies are available against many of the substrates. A fast and simple method to generate cell lysates is by applying multiple freeze/thaw cycles as previously demonstrated33. The use of detergents is not recommended as they can interfere with Gzm activity. In Figure 4B, GzmA mediated cleavage of hnRNPA1 in a HeLa cell lysate is demonstrated in an immunoblot using an anti-hnRNPA1 antibody, as well as an anti-Β-actin antibody as loading control.
For cleavage assays (or cytotoxicity assays) in intact cells, the availability of an additional delivery molecule is necessary (PFN or Streptolysin O, SLO, for mammalian cells; GNLY or other antimicrobial peptides for prokaryotic cells). Cleavage assays in intact cells are most challenging as the concentration of the delivery molecules is critical and needs extensive fine tuning. Introducing these complex protocols of apoptosis assays is beyond the scope of this paper. Here, we may only refer to the vast body of literature, as examples12,13.
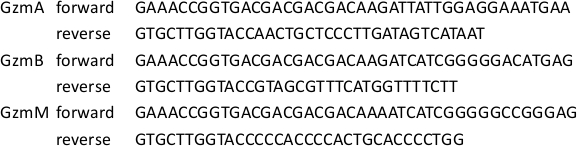
Table 1: Gzm Cloning Primer Sequences.
The primer sequences that were used to clone GzmA, GzmB and GzmM are indicated. Forward primers were designed to introduce an EK site before the N-terminus of the active protease (see Figure 1)

Table 2: Stock Solutions and Buffer Compositions.
The recipes of the most important stock solutions and buffers are indicated.
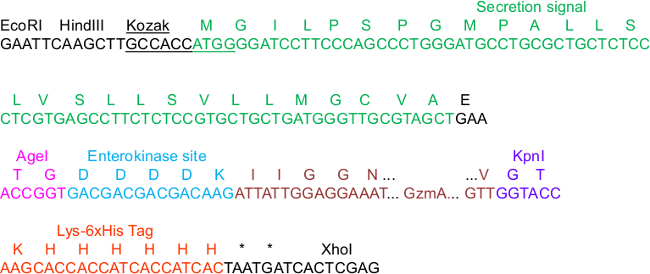
Figure 1. pHLsec Sequence Critical for Construct Cloning.
In the sequence, a few elements in the vector are highlighted that are important for cloning, secretion, IMAC purification and activation of the proteases: The Kozak consensus and secretion signal sequences, the AgeI and KpnI restriction sites, as well as the C-terminal Lys_6xHis tag. We exemplified GzmA as the insert that is N-terminally fused to an enterokinase (EK) site. For the full map of the complete vector backbone, we refer to the supplementary information in23.
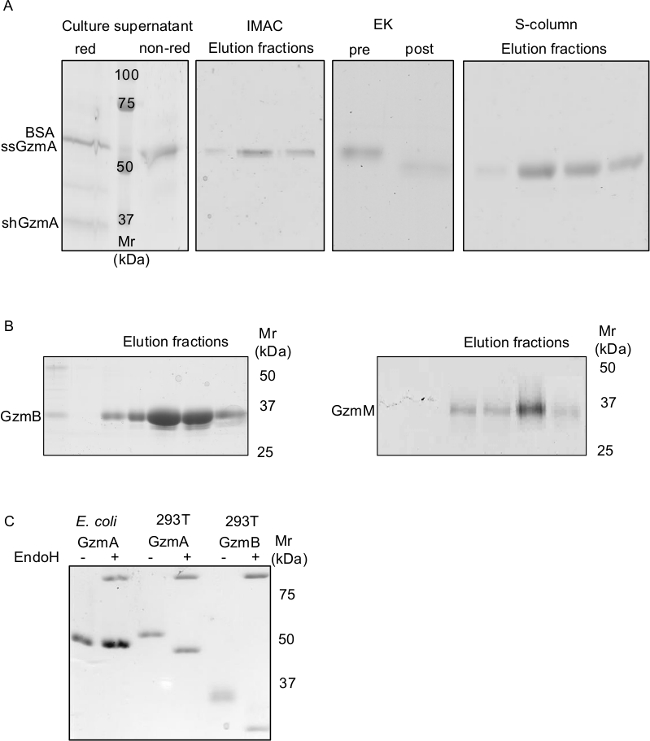
Figure 2. Expression, Purification and Activation of Fully Glycosylated Human Gzms from HEK293T Cells.
A) shows a series of Coomassie-stained, non-reducing SDS-PAGE gels demonstrating the whole GzmA production process from its secretion in the supernatant to first Nickel-IMAC purification, EK treatment and final polishing via cation exchange chromatography. The culture supernatant was run on SDS-PAGE under reducing (red) and non-reducing (non-red) conditions. GzmA is a homodimer (ssGzmA) that is stabilized by a disulfide bond. The GzmA homodimer (~60 kDa) runs close to contaminating BSA (~66 kDa). Under reducing conditions the GzmA monomer (shGzmA) appears as a ~34 kDa band. To demonstrate the purity of the final GzmB and GzmM preparations, representative Coomassie-stained gels under non reducing conditions are shown in B. Bacterially (E. coli) generated GzmA as well as GzmA and GzmB produced in HEK293T cells were treated with EndoH and analyzed by non-reducing SDS-PAGE and Coomassie staining. Glycosylation of the Gzms produced in HEK293T cells is demonstrated by a mobility shift after EndoH treatment as compared to non-glycosylated GzmA produced in bacteria (C).
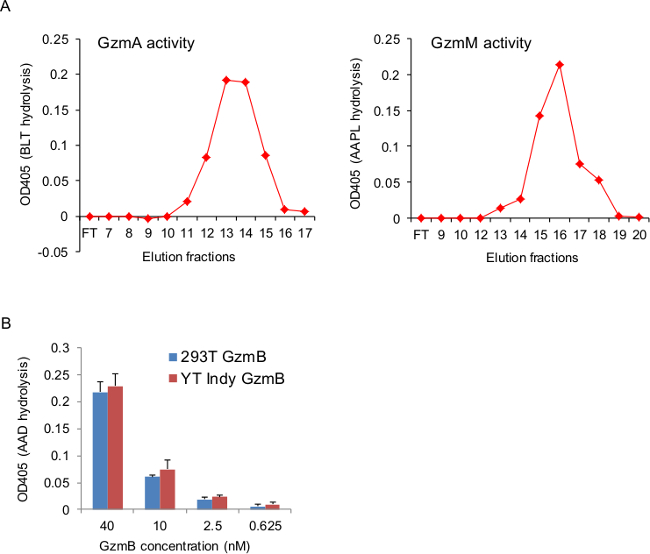
Figure 3. Recombinant Gzms Hydrolyze Synthetic Peptides.
A), small samples of the elution fractions from the final S column were tested for GzmA and GzmM activity in chromogenic assays using BLT and AAPL, respectively, as substrates. Graphs show peptide cleavage that was indicated as an OD increase at 405 nm. The activity peak fractions correlated with the protein peak in the elution as indicated in UV absorbance and SDS-PAGE analysis (Figure 2A and B). B), recombinant 293T and native GzmB (prepared as described12) at indicated concentrations were incubated in presence of the chromogenic substrate AAD. GzmB activity was indicated as an OD change at 405 nm. Graph shows the mean ± SEM of our most recent Gzm preparations that were tested in triplicates.
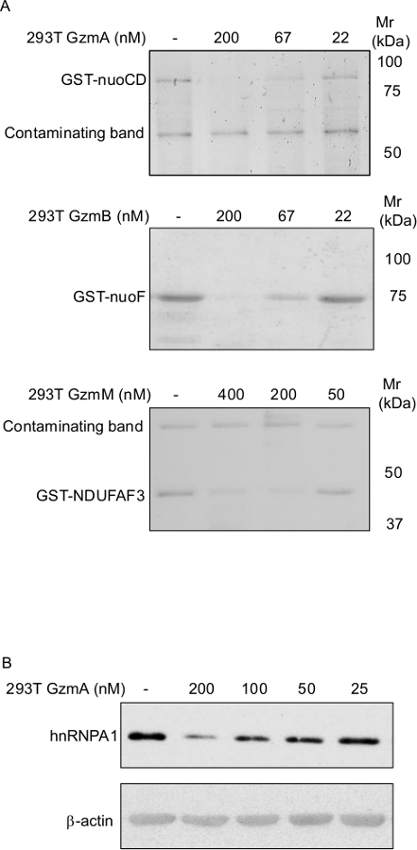
Figure 4. Recombinant Gzms Cleave Protein Substrates.
A) Recombinant GST-tagged, bacterial respiratory chain proteins nuoCD and nuoF, as well as the mammalian respiratory chain protein NDUFAF3, were treated with indicated concentration of indicated Gzms for 15 min at 37 °C. 1 μg of purified proteins in 20 μl reactions were used. Cleavage was analyzed by SDS-PAGE and Coomassie staining. B) HeLa cell lysate (at a protein concentration of ~1 mg/ml) was incubated with indicated concentrations of recombinant GzmA for 30 min and analyzed by immunoblotting using anti hnRNPA1 and Β-actin antibodies.
