This method allows the detection of individual adhesive endothelial cells within a population. For example, while a monolayer of un-irradiated endothelial cells retained a few sporadic HL-60 monocytes following incubation and washing, endothelial monolayer at 7 days post-10Gy irradiation prior to incubation with monocytes were bound by monocytes in clusters around individual endothelial cells (Figure 1). Although this phenomenon is readily observable under phase contrast microscopy, fluorescence microscopy reveals an even clearer image of the monocyte clusters.
After performing the described adhesion assay, it is possible to proceed to staining the cells for membrane, cytoplasmic or nuclear (Figure 2) proteins using appropriate antibodies with standard immunofluorescence methods. Furthermore, enzyme-based assay such as senescence-associated beta-galactosidase (Figure 3) can also be performed.
Since each monocyte cluster corresponds to an individual endothelial cell, enumeration of the clusters will reveal the actual number of adhesive endothelial cells within the monolayer (Figure 4), and hence the percentage of adhesive endothelial cells within the population (Table 1, Figure 6). This is the only method to date that allows such quantification of endothelial cell adhesiveness. It is noteworthy that the endothelial cells used in the experiments here are contact-inhibited and do not increase in number after achieving confluence. This eliminates the potential complexity posed by increased endothelial cell number at later time points. If desired, monocytes attached to endothelial monolayers can be dislodged by 0.5% trypsin-0.2% EDTA solution (instead of fixation in 3.9) and their fluorescence measured using a plate reader, as is the case in contemporary methods (Figure 5).
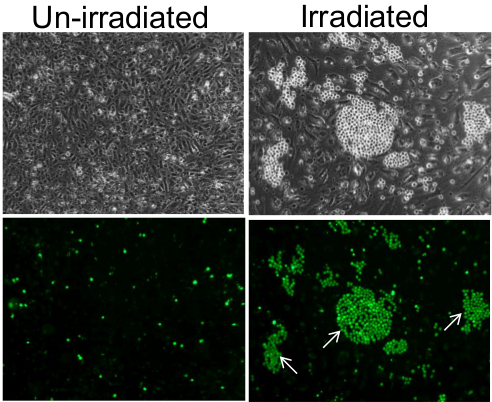
Figure 1. Adhesion of monocytes on monolayer of endothelial cells. On un-irradiated endothelial cells, monocytes adhered as individual cells in a sporadic and random manner (left panels) and in clusters on individual endothelial cells of the 7 days post-10 Gy irradiated monolayer (right panels). Lower panels are fluorescence images of the top panels, confirming that the small and bright spherical cells visible under phase contrast are indeed monocytes that were pre-labeled with Cell-Tracker Green. Some of the monocyte clusters are indicated by white arrows. Images were taken using a 10X objective. Please click here to view a larger version of this figure.
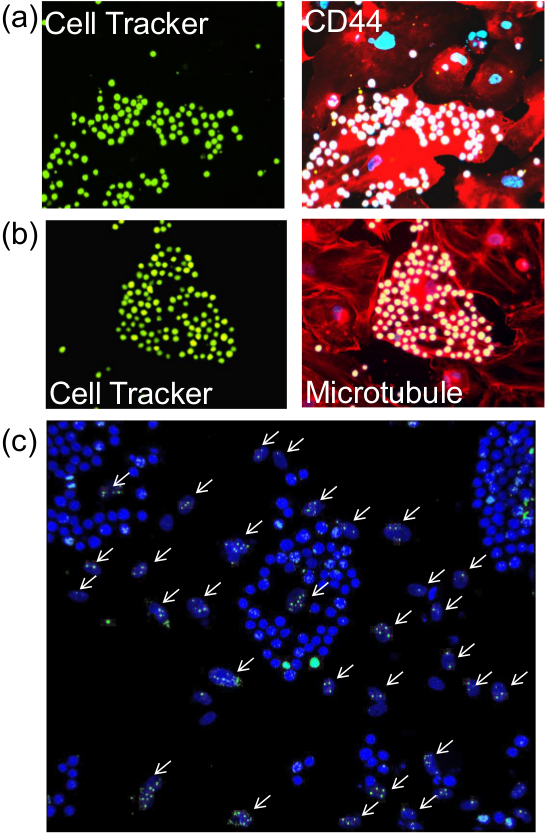
Figure 2. Staining of endothelial cells for proteins post-adhesion assay. After performing the described adhesion assay, cells on the glass coverslips were subjected to immunofluorescence for the detection of (A) membrane protein (CD44), b) cytoplasmic protein (Tubulin) or c) nuclear protein (γ-H2AX). Left panels of (A) and (B) (seen with 20X objective) show monocytes pre-labelled with Cell Tracker Green and the similar image on the right panels reveal CD44 and microtubules (red) and DAPI-stained nuclei (blue). White arrows in (C) point to irradiated endothelial cell nuclei that were stained with antibodies against γ-H2AX. Monocyte nuclei which are stained a brighter blue by DAPI are easily distinguished from the oval shaped nuclei of the endothelial cells. Images were taken with 20X objective. Please click here to view a larger version of this figure.
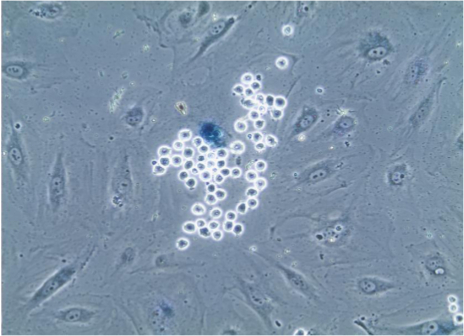
Figure 3. Senescence-associated beta-galactosidase staining of endothelial cells. After adhesion assay, cells on coverslips were subjected to staining for senescence-associated beta-galactosidase which causes lysosomes of senescent cells to turn blue, as is evident in the endothelial cell that is selectively bound by numerous monocytes, which are easily identified due to their small size and spherical shape. Image seen through 20X objective. Please click here to view a larger version of this figure.
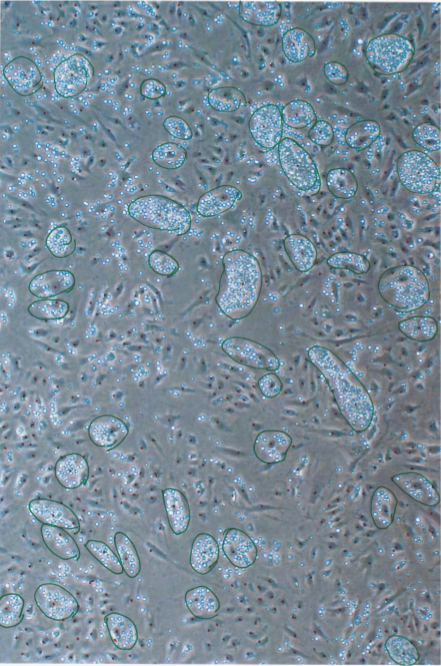
Figure 4. Enumeration of monocyte clusters on individual endothelial cells. After adhesion assay, images of the cells were taken from several different positions and monocyte clusters identified and circled (in green). A cluster was defined as an agglomeration of 10 or more monocytes on an endothelial cell. The number of monocyte clusters and number of 12 days post-irradiated endothelial cells (Dotted in red) in the image were counted and percentage of adherent endothelial cells calculated as shown in Table 1. Image taken through 10X objective. Please click here to view a larger version of this figure.
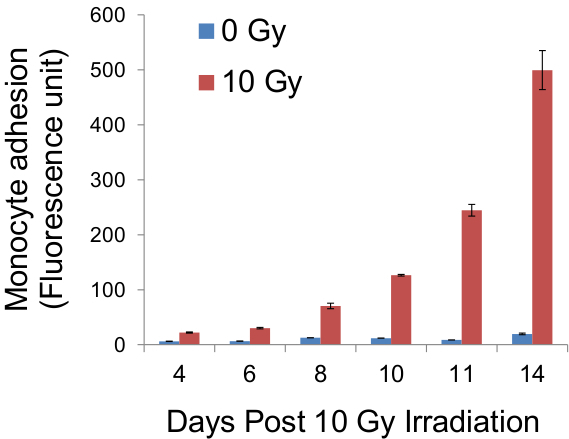
Figure 5. Quantification of fluorescence of adherent monocytes. Following adhesion assay, cells on glass coverslips were trypsinised and the fluorescence of monocytes pre-labelled with CellTracker Green was measured, using a fluorescence plate reader, as an indirect indicator of the adhesiveness of the endothelial monolayer. Results from such measurements at stated days post-irradiation were obtained and platted on the graph above.
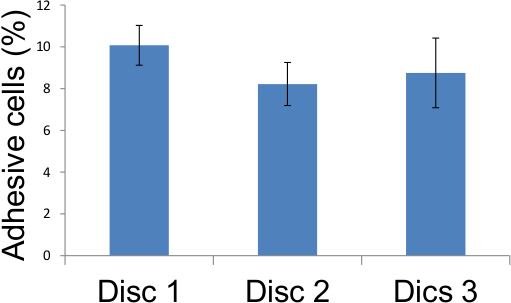
Figure 6. Representation of adhesion assay scores from Table 1. Adhesive endothelial cells in five different locations on three glass coverslips were scored as described in Figure 4 and the results tabulated above demonstrating that 12 days after 10 Gy irradiation, 8 to 10 percent of irradiated endothelial cells became adhesive.
| Percentage of adhesive endothelial cells |
|||
| Disc 1 | Disc 2 | Disc 3 | |
| Field 1 | 8.65 | 8.29 | 8.21 |
| Field 2 | 10.44 | 7.27 | 7.21 |
| Field 3 | 9.63 | 9.05 | 8.33 |
| Field 4 | 11.11 | 7.09 | 8.41 |
| Field 5 | 10.55 | 9.4 | 11.61 |
| Average | 10.07 | 8.22 | 8.75 |
Table 1. Adhesion assay scores. Adhesive endothelial cells in five different locations on three glass coverslips were scored as described in Figure 4 and the results tabulated above demonstrating that 12 days after 10 Gy irradiation, 8 to 10 percent of irradiated endothelial cells became adhesive.
