After finishing the extracellular flux assay and cell quantification, data can be normalized for cell counts and analysed. This will typically yield ECAR and OCR plots as shown in Figure 1A and Figure 1B.
After the injection of glucose (A), the increase in ECAR represents the glycolysis rate. The additional increase in ECAR after ATP synthase inhibition with oligomycin (B) provides information about the glycolytic reserve and capacity (Figure 1A). When analysing the OCR values, oligomycin injection (B) allows calculation of the oxygen consumption used for mitochondrial ATP synthesis. FCCP (C) uncouples mitochondrial respiration and the corresponding OCR measurements yield data about the maximal and spare respiratory capacity. Finally, injection of rotenone (Rot) and antimycin A (AA) block mitochondrial complex I and III and the residual OCR represents the non-mitochondrial oxygen consumption (Figure 1B).
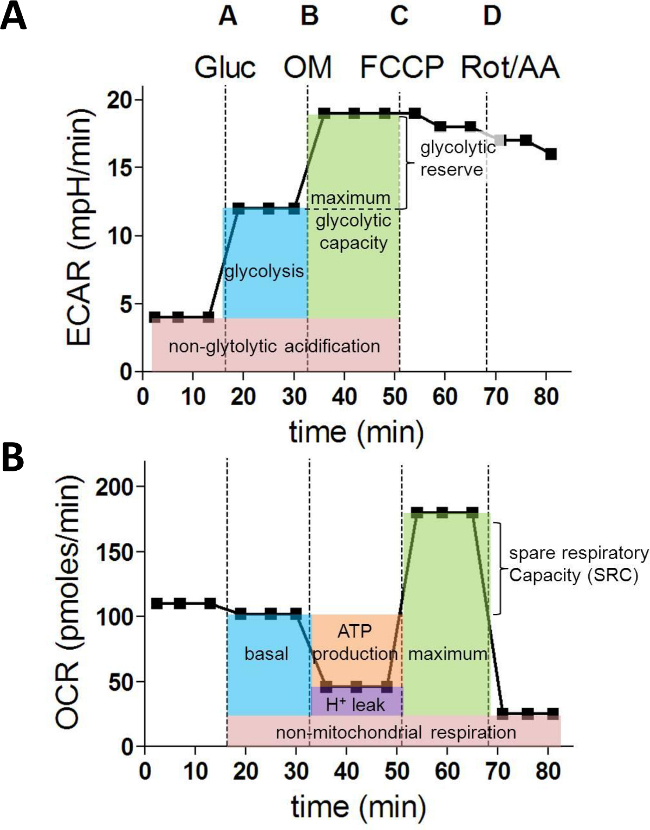
Figure 1: Metabolic parameters derived from a XF Extracellular flux assay. (A) Following parameters of cellular glycolysis are calculated from the ECAR values (in mpH/min): glycolysis, maximum glycolytic capacity and glycolytic reserve. (B) OCR measurements (in pMoles/min) are used to calculate the next fundamental parameters of mitochondrial function: basal respiration, ATP production, proton leak, maximum respiration and spare respiratory capacity. Gluc = glucose; OM = oligomycin A; FCCP = Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; AA = antimycin A; Rot = Rotenone Please click here to view a larger version of this figure.
After the run, ECAR and OCR measurements 1 till 15 for each well can be exported to Excel and the following glycolytic parameters can be calculated from the ECAR measurements as follows:
Non-glycolytic acidification = avg. ECAR(1,2,3)
Glycolysis = avg. ECAR(4,5,6)– avg. ECAR(1,2,3)
Maximum glycolytic capacity = avg. ECAR(7,8,9)– avg. ECAR(1,2,3)
Glycolytic reserve= avg. ECAR(7,8,9) – avg. ECAR(4,5,6)
From the OCR rates, the next metabolic characteristics can be determined:
Non-mitochondrial respiration = avg. OCR(13,14,15)
Basal respiration = avg. OCR(4,5,6) – avg. OCR(13,14,15)
ATP production = avg. OCR(4,5,6) – avg. OCR(7,8,9)
Proton leak = avg. OCR(7,8,9) – avg. OCR(13,14,15)
Maximum respiration = avg. OCR(10,11,12) – avg. OCR(13,14,15)
Spare respiratory capacity (SRC) = avg. OCR(10,11,12) – avg. OCR(4,5,6)
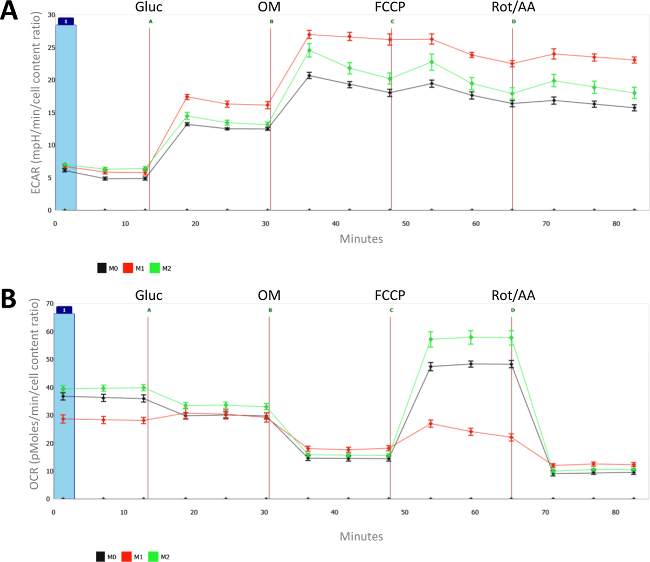
Figure 2: Metabolic characteristics of naïve (M0), LPS- (M1) and IL-4- (M2) polarized macrophages. For each measurement, the mean and standard error of the mean (SEM) of 8 individual wells is presented. (A) Extracellular acidification rates (ECAR, in mpH/min) and (B) oxygen consumption rates (OCR, in pMoles/min) are measured for differentially polarized (M1 and M2) and naïve macrophages (M0). All measurements were normalized for cell count (as measured with CyQUANT) and the average cell count of the naïve macrophages was set at 1. Injection A = glucose; B = oligomycin; C = FCCP; D= antimycin A + Rotenone. Please click here to view a larger version of this figure.
As shown in Figure 2A, macrophage activation with LPS induces increased glycolytic metabolism. The differences between LPS and IL-4-treated macrophages are even more apparent when looking at oxygen consumption rates (OCR) in Figure 2B. Indeed, while the maximal oxidative metabolism is highly suppressed in M(LPS), IL-4 induces basal and especially maximal respiration in M2 macrophages. One should note that increased glycolytic is rather unique to M(LPS) macrophages and is not necessarily a characteristic of M1 macrophages since M(IFNγ) macrophages do not display enhance glycolysis.
Overall, this extracellular flux analysis demonstrates that IL-4-induced M2 macrophages are characterized by enhanced oxidative metabolism and especially a high spare respiratory capacity (SRC), whereas LPS-activated macrophages display enhanced glycolytic metabolism and do not possess any spare respiratory capacity.
