Using this method, yeast PtdInsPs were metabolically labelled with 3H-myo-inositol. After labeling, the phospholipids were precipitated with perchloric acid, followed by phospholipid deacylation and extraction of the water-soluble Gro-InsPs (Figure 1). At this stage, it is important to quantify the total radioactive signal associated with the extracted Gro-InsPs by liquid scintillation to ensure sufficient signal-to-noise ratio for the very low-abundance PtdInsPs like PtdIns(3,5)P2; a total of 5-10 million CPMs should be injected. Since yeast cells exhibit only four PtdInsPs, resolution can be achieved with a 1 hr elution method as described (Figure 2).
Here, wild-type, atg18Δ and vac14Δ yeast mutants were labelled and processed to analyse changes in PtdIns(3,5)P2, the least abundant of the PtdInsPs in yeast16,19,27. As observed in Figure 4A-C, the elution profile for all three strains produced 3H-associated signal peaks. The parental Gro-Ins peak elutes first (8-9 min; shown in Figure 3, but not in Figure 4 since Gro-Ins overshadows the signal of the phosphorylated species), followed by Gro-Ins3P (~18 min), Gro-Ins4P (~20 min), Gro-Ins(3,5)P2 (~29 min) and Gro-Ins(4,5)P2 (~32:00 min), which respectively corresponds to the acylated phospholipids PtdIns, PtdIns3P, PtdIns4P, PtdIns(3,5)P2 and PtdIns(4,5)P2. There are a couple of additional minor peaks at 4 min and one that tends to immediately follow the parent Gro-Ins (10 min); these likely represent inositol and a non-glycerated inositide phosphates (Figure 3). The elution pattern is consistent and characteristic, though the exact time may vary when employing a different HPLC system and/or a new column.
The peak associated with PtdIns(3,5)P2 (Figure 4, red arrow) undergoes the most dramatic change between wild-type, atg18Δ and vac14Δ yeast strains. Relative to wild-type cells (A), there is a very large Gro-Ins(3,5)P2-associated signal in atg18Δ cells and a smaller peak in vac14Δ yeast cells. In order to quantify the total radioactive signal associated with each peak, the counts were integrated under the area of the peak (Figure 3). In addition, since absolute radioactive signal varies between samples, the parental Gro-Ins species was used as an internal control by normalizing against it (one can also choose to normalize against total counts). By doing this, the data suggest that PtdIns(3,5)P2 constitutes only 0.07% of PtdIns in wild type yeast cells, while PtdIns(3,5)P2 corresponds to 1.2% of the parental PtdIns in atg18Δ cells, or a dramatic 17-fold increase in PtdIns(3,5)P2 relative to wild-type cells (Figure 4A, B and D). In comparison, PtdIns(3,5)P2 levels was only 0.01% of the PtdIns signal in VAC14-deleted yeast cells, an 85% loss in PtdIns(3,5)P2 (Figure 4C and D). Overall, these measurements are in agreement with published results showing that PtdIns(3,5)P2 is about 0.1% of PtdIns in wild-type cells, 90% reduced in vac14D, and 10 to 20-times higher in atg18D cells relative to wild-type cells27,29.
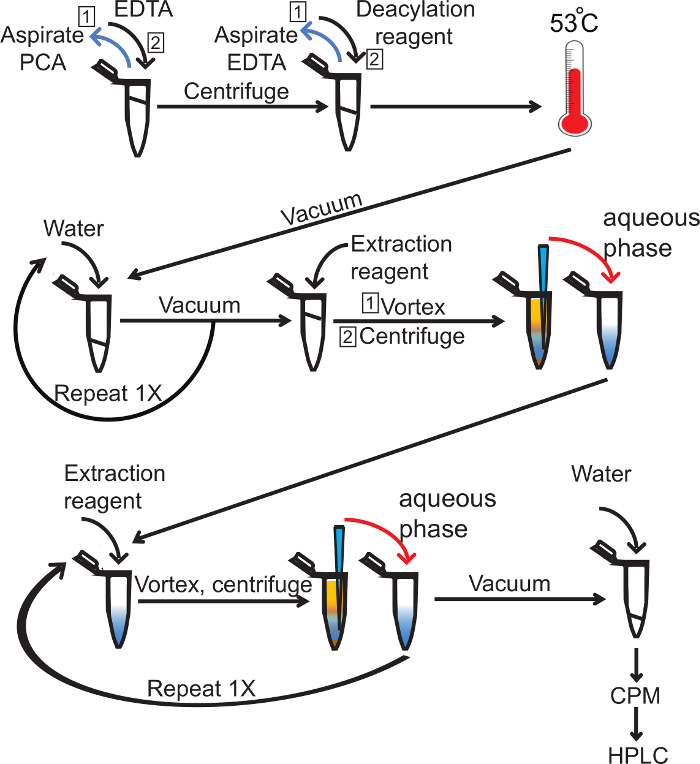
Figure 1. Deacylation and extraction of 3H-Gro-InsP. Radiolabeled lipid precipitate in perchloric acid is washed with aqueous solution of EDTA. This is then aspirated and deacylation reagent added and incubated at 53 °C. The deacylation reaction mixture is then vacuum dried and washed with water twice, followed by the addition of extraction reagent. This is then vortexed, centrifuged and the aqueous phase is collected. The extraction step is repeated three times total to remove organic and insoluble contaminants. The water-soluble fraction (blue), which includes 3H-Gro-InsP, is then vacuum dried and rehydrated with 60 µl of water. The amount of radioactivity is determined by liquid scintillation (CPM) and equal counts across samples are loaded into the HPLC. Please click here to view a larger version of this figure.
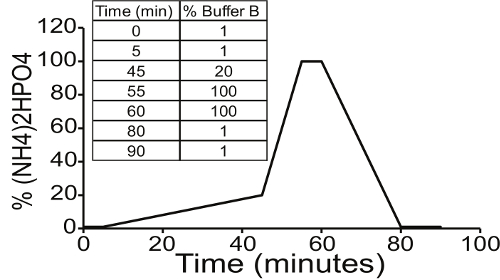
Figure 2. Gradient of ammonium phosphate buffer for elution of 3H-Gro-InsP. Resolution of Gro-InsP by strong anion exchange chromatography depends on the application of ammonium phosphate dibasic, pH 3.8 (Buffer B). The choice of gradient depends on the mixture of Gro-InsP species available for separation. Protocol A is used for yeast samples, which possess only four PtdInsPs. Resolution of each of the four peaks is sufficient with a rapid increase in (NH4)2HPO4 concentration. Please click here to view a larger version of this figure.
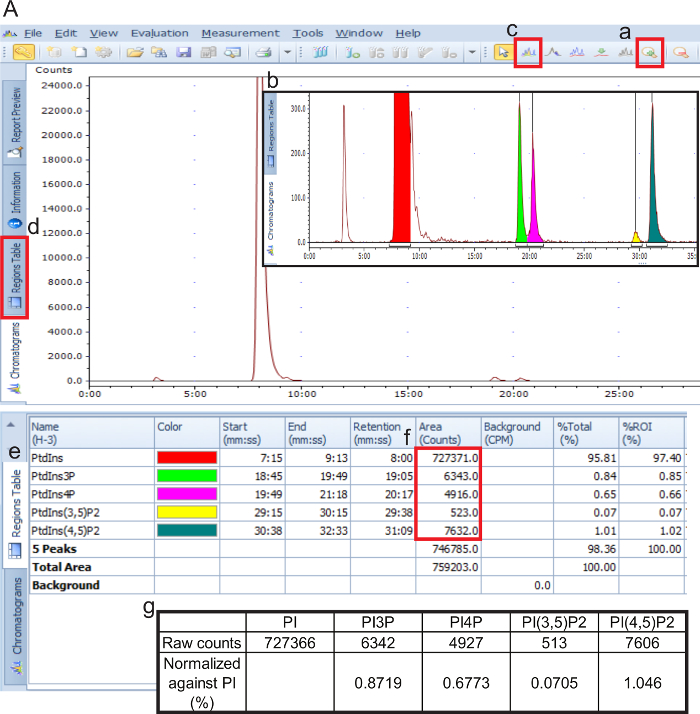
Figure 3. Guided instruction for data analysis of PtdInsP abundance. Data files are loaded on the Laura software and evaluated visually using the chromatograph (open by default). Using the "zoom in" tool (a), magnify the peaks (b). Select distinct peaks on the chromatograph using the "add ROI" tool (c). In the "region table" tab (d), all resulting information from the highlighted regions of interest is displayed as a single table (e). Export the raw counts of each peak (f) and normalize each PtdInsP species against the parental phosphatidylinositol (g) or against total counts. Please click here to view a larger version of this figure.
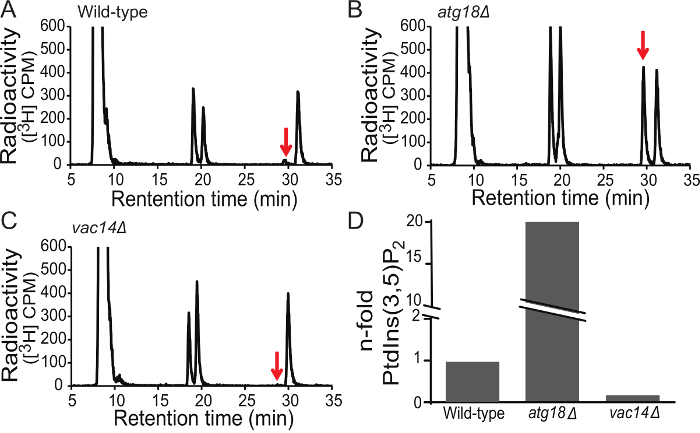
Figure 4. PtdIns(3,5)P2 levels in wild-type and mutant yeast cells. Representative chromatographs are shown for the flow scintillation of the extracted 3H-Gro-InsPs from wild-type (A), atg18Δ (B), and vac14Δ (C) yeast strains using Protocol A. The raw counts from the regions of interest corresponding to Gro-Ins(3,5)P2 (arrow) are extracted from the chromatograph and background subtracted. The resulting values are compared to wild-type and expressed as fold-change in Gro-Ins(3,5)P2 levels (D). The levels and changes in Gro-Ins(3,5)P2 are interpreted to show levels and changes in the original PtdIns(3,5)P2. Please click here to view a larger version of this figure.
| Inositol-free media (IFM) |
| 27.8 mM (NH4)2SO4 |
| 2% Glucose |
| 0.01 mM KH2PO4 |
| 1X Amino acids |
| 1X LPSM |
| 1X Trace elements |
| 5X Vitamins |
| 10X Low phosphate and sulfate media (LPSM) |
| 9 mM CaCl2 |
| 17 mM NaCl |
| 67 mM KCl |
| 63 mM MgCl2 |
| 1,000X Trace elements |
| 8 mM Boric acid |
| 250 µM CuSO4 |
| 602 µM KI |
| 1.23 mM FeCl3 |
| 2.65 mM MnSO4 |
| 971 µM Na-molybdate |
| 2.48 mM ZnSO4 |
| 500X Vitamins |
| 4 µM Biotin |
| 839 µM Ca-pantothenate |
| 2.27 µM Folic acid |
| 1.62 mM Niacin |
| 729 µM p-aminobenzoic acid |
| 973 µM Pryidoxine-HCl |
| 266 µM Riboflavin |
| 593 µM Thiamine-HCl |
Table 1. Composition of the yeast inositol-free media. Yeast cells are radiolabelled with IFM as the base media to be supplemented with 3H-myo-inositol. Prepare a 100 ml solution of IFM using the solutions indicated, filter sterilize using a bottle top 0.22 µm vacuum filter and store at RT. To prepare low phosphate and sulfate media (LPSM), generate a 100 ml solution using the indicated salts, filter sterilize using a bottle top 0.22 µm vacuum filter and store at RT. Dissolve the indicated salts to prepare a 1 L 1,000x solution of trace elements. Mix thoroughly before use and/or store aliquots at -20 °C. Dissolve the indicated vitamins to prepare a 1 L 500x vitamin solution. Mix thoroughly before use and store aliquots at -20 °C.
