Fresh placentas from term pregnancies were used to isolate human primary villous cytotrophoblasts to conduct the set of experiments presented in the Protocol section. Following their isolation, we first analyzed the purity of cytotrophoblasts through the use of the cytokeratin-7 marker (Figure 1). Cell preparations were thus stained using a monoclonal anti-cytokeratin-7 antibody. Figure 1 represents results from a typical experiment following purification of primary villous cytotrophoblasts, analyzed by standard flow cytometry. Following the density gradient step, >97% cells (in this case, 99%) are positively stained for cytokeratin-7 in standard experiments, thereby demonstrating a very high degree of efficiency in the purification of this cell type.
After their isolation, human primary cytotrophoblasts can be directly used for transfection. Two different protocols were evaluated, i.e., microporation and lipid-based transfection. Both protocols were tested with siRNA specific to Syncytin-2 mRNA and compared to a scrambled siRNA (negative control). Transfection efficiency was next evaluated by Western blot analysis. Results confirmed that Syncytin-2 expression was significantly silenced in cytotrophoblast-derived extracts upon microporation (Figure 2). On the other hand, no significant silencing was noted when siRNAs were transfected by the lipid-based transfection reagent.
We also conducted EMSA experiments on freshly isolated cytotrophoblasts. A radiolabeled probe that originated from a Syncytin-2 promoter region was synthesized. The synthesized probe (termed WT; 5'-CTCTAGGAACACCTGACTGATAAGGGAAAAATGTC-3') has been previously shown to bind CREB-related transcription factors 7. In the presence of nuclear extracts from 24 and 48 hr cultured villous cytotrophoblasts, the Syncytin-2-promoter-derived probe showed the presence of two DNA-protein complexes. Addition of 100x cold WT oligonucleotide competed for the formation of the complex, while no similar competition with excess of a cold unrelated oligonucleotide (mouse Neural Crest Enhancer 2; 5′-GATCCTGTGATTTTCGTCTTGGGTGGTCTCCCTCG-3') was observed, confirming the specificity of both signals (Figure 3).
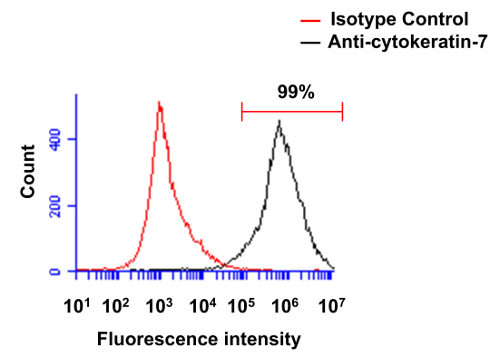
Figure 1. Evaluation of the purity of primary villous cytotrophoblasts through flow cytometry. Freshly isolated preparations of villous cytotrophoblasts were first permeabilized and then labeled with isotype control antibody (red line) or monoclonal antibody specific for cytokeratin-7 (black line). Cells were analyzed by flow cytometry. Please click here to view a larger version of this figure.
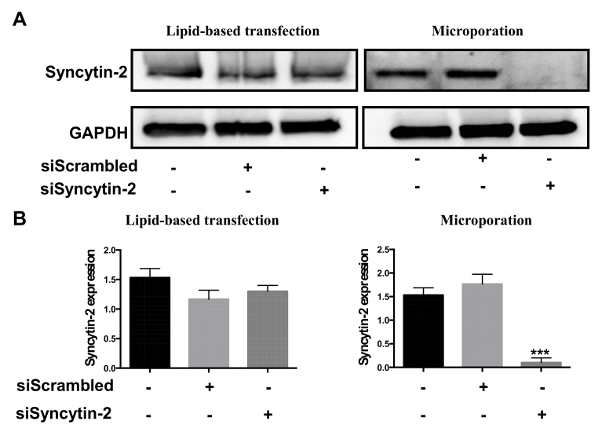
Figure 2. Comparison of siRNA transfection efficiencies between lipid-based transfection and microporation. Freshly isolated villous cytotrophoblasts were transfected with 300 ng of Syncytin-2-specific siRNA vs. a scrambled control siRNA using the lipid-based reagent or microporation. (A) Western blot analyses were performed on cellular extracts from each transfection condition using anti-Syncytin-2 and anti-GAPDH antibodies. (B) Syncytin-2 protein levels from Western blot analyses were quantified in terms of band intensity following normalization with corresponding GAPDH signals (Error bars are defined as SD, ***p<0.001). Please click here to view a larger version of this figure.
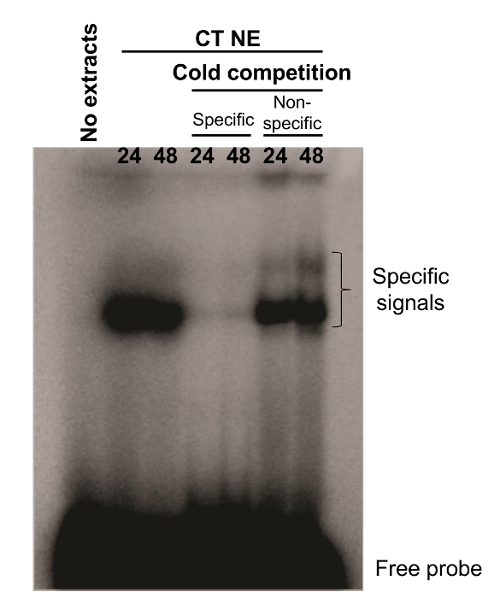
Figure 3. DNA-protein complexes identified by EMSA analysis. Nuclear extracts from primary villous cytotrophoblasts cultured for 24 or 48 hr were incubated with a WT probe corresponding to a promoter region of Syncytin-2 with or without excess (100x) of specific or nonspecific cold oligonucleotides. DNA-protein complexes were separated upon migration on a native gel. Specific signals and free probes are indicated on the right side of the gel. WT probe incubated in the absence of nuclear extract was also used as a negative control. CT NE = cytotrophoblast nuclear extracts. Please click here to view a larger version of this figure.
| % of Percoll final | Volume of Percoll (90%) (ml) | Volume of HBSS (ml) |
| 70 | 2.33 | 0.67 |
| 65 | 2.17 | 0.83 |
| 60 | 2.00 | 1.00 |
| 55 | 1.83 | 1.17 |
| 50 | 1.67 | 1.33 |
| 45 | 1.50 | 1.50 |
| 40 | 1.33 | 1.67 |
| 35 | 1.17 | 1.83 |
| 30 | 1.00 | 2.00 |
| 25 | 0.83 | 2.17 |
| 20 | 0.67 | 2.33 |
| 15 | 0.50 | 2.50 |
| 10 | 0.33 | 2.67 |
| 5 | 0.17 | 2.83 |
Table 1. 5%-70% polyvinylpyrrolidone-coated silica gradient setting.
| Buffer | Composition | Comments/Description |
| PBS 1x | 0.9 mM CaCl2, 2.7 mM KCl, 1.5 mM KH2PO4, 0.5 mM MgCl2, 136.9 mM NaCl and 0.8 mM Na2HPO4 | |
| RIPA Buffer | 150 mM NaCl, 1.0% NP-40 or 0.1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS (sodium dodecyl sulfate), 50 mM Tris-HCl pH 8.0 | |
| Laemmli Sample Buffer | 4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, 0.125 M Tris-HCl, pH 6.8 | |
| 10x running buffer | 25 mM Tris base, 190 mM glycine, 0.1% SDS | |
| 10x transfer buffer | 25 mM Tris base, 192 mM Glycine | |
| TBST 1x buffer | 10 mM Tris-HCl, 15 mM NaCl, 0.05% Tween 20 at pH 7 | Mix well and filter. Failure to filter can lead to “spotting” of the membrane. |
| Blocking buffer | 5% milk or BSA (bovine serum albumin) added to TBST 1x buffer | |
| TE Buffer | 10 mM Tris-HCl (pH 8.0), 1 mM EDTA | |
| Annealing buffer | 1 M NaCl, 50 mM Tris-HCl pH 7.5, 100 mM MgCl2, 0,2 mM EDTA, 1 mM DTT | |
| 5x Gel Shift Binding Buffer | 20% glycerol, 5 mM MgCl2, 2.5 mM EDTA, 2.5 mM DTT, 250 mM NaCl, 50 mM Tris-HCl (pH 7.5) and 0.25 mg/ml poly(dI-dC). | |
| Loading buffer | 250 mM Tris-HCl (pH 7.5), 0.2% bromophenol blue and 40% glycerol | |
| TBE 5x | Dissolve 54 g of Tris Base and 27.5 g of boric acid in 1 L deionized water, which includes 20 ml 0.5 M EDTA, pH 8 | |
| 4% native gel | TBE 5x buffer | 6.0 ml |
| 29:1 acrylamide/bisacrylamide (30% w/v) | 10 ml | |
| 40% acrylamide (w/v) | 0.75 ml | |
| 100% glycerol | 1.5 ml | |
| Distilled water | 41.5 ml | |
| Ammonium persulfate 25% | 250 ml | |
| TEMED | 75 ml | |
| 4% Stacking Gel (6 ml) for SDS-PAGE | ddH2O | 4.1 ml |
| 30% Acrylamide | 1.0 ml | |
| 1.0 M Tris | 0.75 ml | |
| 10% SDS | 60 μl | |
| 10% APS | 60 μl | |
| TEMED | 6 μl | |
| 12% Separating Gel (10 ml) for SDS-PAGE | ddH2O | 3.3 ml |
| 30% Acrylamide | 4.0 ml | |
| 1.5 M Tris | 2.5 ml | |
| 10% SDS | 100 μl | |
| 10% APS | 100 μl | |
| TEMED | 4 μl |
Table 2. Composition of buffers.
