VLP yields were variable by viral antigen construct design. In this protocol, we have demonstrated use of insect and mammalian cells for production of SVP or VLPs in supernatant and purification by ultracentrifugation. Four subtypes of DENV prM/E structural gene expression cassettes were used to construct the versions of DENV SVPs (demarcated as 1-4) in Table 1 and demonstrate a range between 1.1-2.6 mg of total protein in 0.6 ml volumes. For VLPs that require a three gene constructs, we found the optimized DNA transfection ratio of 1:1:2 for HA:NA:Gag respectively to synthesize influenza VLPs; in contrast, a single plasmid expressing multiple chikungunya viral proteins was adequate for the generation of recombinant baculovirus and mammalian CHIK VLP. Additionally, a dual-plasmid transfection procedure is also possible with mammalian cells, as such with RSV F and Gag transfected at a 1:1 ratio, and yields approximately 0.01 mg/ml of total cell culture supernatant. As shown in the Table 1, CHIK SVP yield from Sf9 cells can range between 0.008-0.016 mg total protein/ml of supernatant volume, while production through mammalian 293T yields a 10-fold reduction of protein. Between the two versions of H3N2 VLPs using different wild-type, full-length HA plasmids, the H3N2 VLP Sample 2 had a reduced yield of both total protein and specific HA content. Other sample VLP HA quantities are shown in Figures 2 and 3 and illustrate how HA and ELISA are used to estimate surface content on the VLPs. As with a conventional direct ELISA method, a standard curve is generated using known concentrations of a recombinant HA to compare ELISA reactivity to sample H3 VLPs loaded onto an ELISA plate. ELISA or similar immunoassay are recommended for quantification of VLPs that have readily available monoclonal antibodies with known crossreactivity to a well-conserved epitopes, but may not be available for all antigens. RSV F is well-characterized, therefore an ELISA using monoclonal anti-RSV F antibody is also applicable for quantification of surface F on pelleted RSV F VLPs. In addition to antigen functionality and antigen recognition by specific antibodies, VLP preparations can be structurally assessed by electron microscopy. Figure 4 is a representative image showing that HA (Gag-core) influenza VLPs are successfully expressed, assembled, and purified as VLPs.
| Description | Culture Vol (ml) | Harvesta | Total Volume | Total Protein Yield (mg) | Yield (mg)/Vol (ml) | Specific antigen content |
| DENV1 SVP | 293T186 | 4 d | 0.6 ml | 1.115 | 0.006 | n.d. |
| DENV2 SVP | 293T/186 | 4 d | 0.6 ml | 2.5 | 0.0134 | n.d. |
| DENV3 SVP | 293T/186 | 3 d | 0.6 ml | 1.536 | 0.0083 | n.d. |
| DENV4 SVP | 293T/186 | 4 d | 0.6 ml | 2.613 | 0.014 | n.d. |
| CHIK VLP | Sf9/186 | 3 dpi | 0.6 ml | 1.503 | 0.0081 | n.d. |
| CHIK VLP | Sf9/500 | 4 dpi | 2.0 ml | 8.276 | 0.0166 | n.d. |
| CHIK VLP | 293T/186 | 3 d | 0.6 ml | 0.296 | 0.0016 | n.d. |
| H3N2 VLP 1 | 293T/216 | 3 d | 0.6 ml | 1.25 | 0.0058 | 1.9 µg HA/μl |
| H3N2 VLP 2 | 293T/216 | 3 d | 0.6 ml | 0.956 | 0.0044 | 0.96 µg HA/μl |
| H3N2 VLP 3 | 293T/216 | 3 d | 0.6 ml | 1.6 | 0.0074 | 1.2 µg HA/μl |
| H3N2 VLP 4 | 293T/216 | 3 d | 0.6 ml | 1.54 | 0.0071 | 0.98 µg HA/μl |
| RSV F VLP | 293T/216 | 3 d | 0.6 ml | 2.3 | 0.0106 | 0.15 µg RSV F/μl |
| a d = days post-transfection, dpi = days post-infection | ||||||
Table 1: Subviral and virus-like particle yields by construct. Representative data of VLP volumes, total protein yield (determined by conventional BCA assay) or specific antigen content (determined by ELISA) is shown among a variety of SVP/VLP constructs. Yields are variable due to difference in SVP/VLP assembly and cell type, and maximum yields may require user optimization. The various versions of either DENV1-4 differ based on serotype, while the CHIK VLPs use the same sequence but preparations differ in either volume or cell culture type. The different versions of H3N2 VLPs (1-4) express four unique HA sequences that share a HA-specific ELISA reactive epitope and were co-expressed with the same NA and Gag core. The RSV F VLP, which is generated from a dual-plasmid transfection of RSV F and Gag, was synthesized and RSV F content was approximated using RSV F specific ELISA assay.
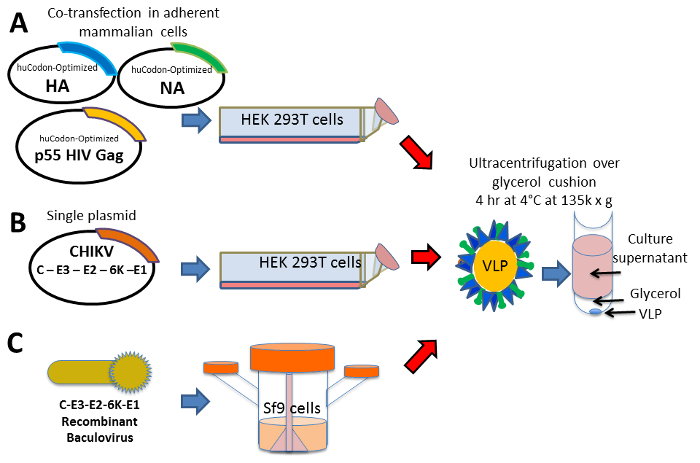
Figure 1: Approach – VLP design, synthesis, and purification. Schematic representation of plasmid gene designs for expression of viral structural proteins that will form VLPs: (A) co-expression of multiple structural proteins by co-transfection of three plasmids in 293T cells, (B) expression of structural proteins from a single gene cassette encoded in a single plasmid in 293T cells, and (C) recombinant baculovirus encoding CHIK structural proteins is used to infect Sf9 cells cultured in spinner flasks to promote high density cell growth in suspension. The VLPs are harvested from the cell culture media, clarified of cell debris, and separated from non-particles via sedimentation by ultracentrifugation on a 20% glycerol cushion. Please click here to view a larger version of this figure.
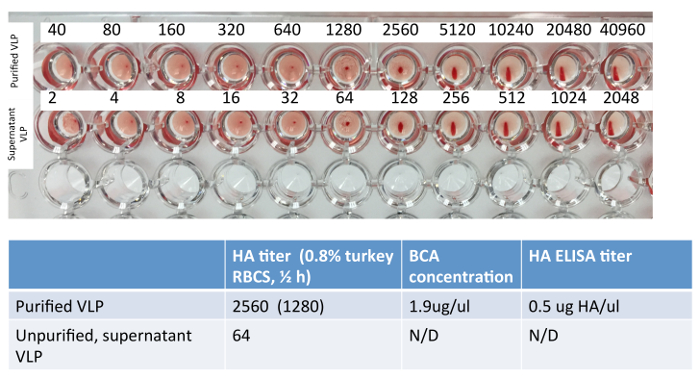
Figure 2: Example Hemagglutination assay of H3 VLPs. A conventional hemagglutination assay was performed on a representative sample of H3N2 VLP and its corresponding clarified supernatant; H3N2 HA assay can be performed using 0.8% turkey red blood cells or guinea pig red blood cells (not shown). Total protein content is estimated using the standard BCA assay on the purified VLP and was not performed or recommended for clarified supernatant. ELISA was performed on the purified VLP and HA content was estimated using a recombinant HA standard of known concentration. Please click here to view a larger version of this figure.
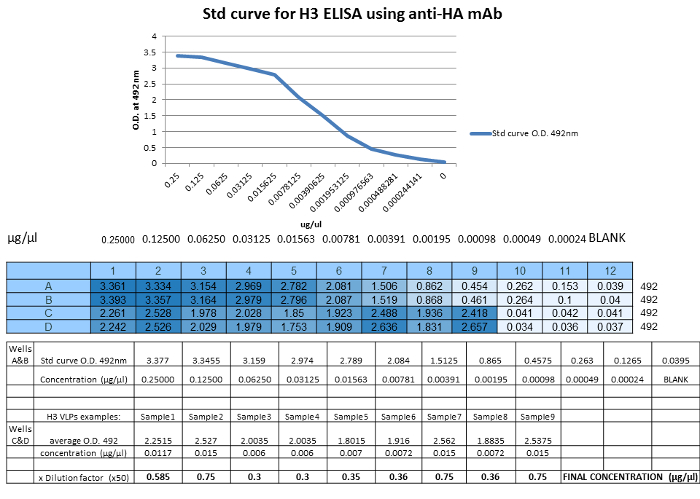
Figure 3: Example H3 ELISA for quantification of H3 content in H3 VLPs. An ELISA plate was coated with serial dilutions of a recombinant HA standard of known concentration and diluted H3N2 VLP samples generated from different HA sequences (Samples 1-9). Correlating the concentration (µg/µl) of the standard to the optical density values obtained at 492 n.m. (O.D.), concentrations are obtained by deducing the x-value that intersects the linear portion of the standard curve. Dilutions of recombinant HA standard and unknown samples were loaded in duplicate in wells A& B or C & D, respectively. The final concentration of the VLP sample is determined by multiplying the concentration by the dilution factor of the VLP sample. Please click here to view a larger version of this figure.
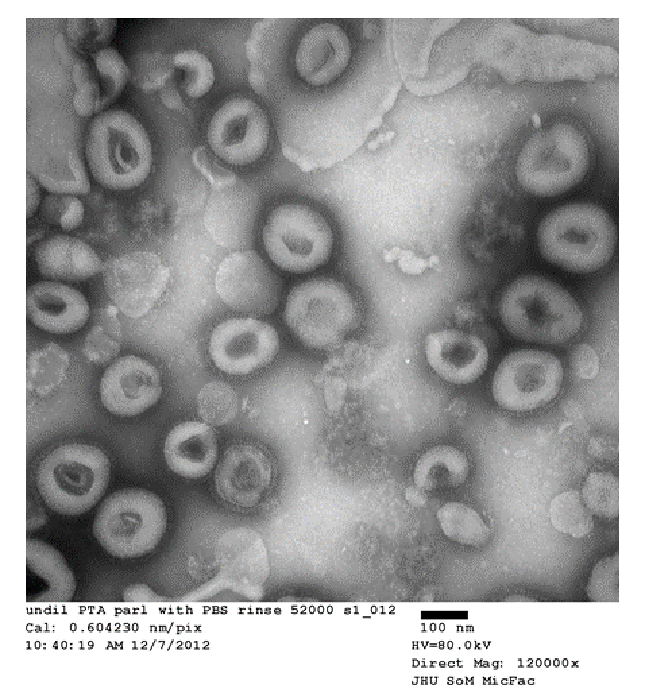
Figure 4: Electron microscopy of HA-VLPs using Gag-core. Representative electron microscopy image of purified influenza HA VLPs. VLPs assemble into spherical particles coated with HA. Please click here to view a larger version of this figure.
