To examine giant lampbrush chromosomes one begins by isolating oocytes from a frog or salamander. Figure 1 shows a group of mature oocytes in a buffered saline solution after removal from the ovary of the frog, Xenopus. Such oocytes remain in good condition for days at room temperature. The nucleus (or germinal vesicle) is then removed from an oocyte with jeweler's forceps, either in a saline solution (Figure 2) or in oil (Figure 3). The nuclear contents of an oil-isolated nucleus can be examined by gently squashing the nucleus and observing by phase contrast or differential interference contrast (DIC) microscopy. To examine the contents of a nucleus that has been isolated in a saline solution, one must first remove the nuclear envelope with jeweler's forceps and allow the contents to settle onto a microscope slide. The preparation is then centrifuged to attach the chromosomes firmly to the slide, after which the chromosomes can be stained with an antibody (Figure 4) or subjected to in situ hybridization. The lampbrush chromosomes of salamanders are much larger than those of frogs (Figure 5). In both cases individual transcription units (genes) can be seen as loops of chromatin projecting laterally from the chromosome axis.

Figure 1: Mature Oocytes from the Frog X. tropicalis. The ovary of a mature female frog contains thousands of oocytes in different stages of maturation. The smallest are the size of somatic cells and have already reached prophase of the first meiotic division. As the oocyte grows, it gradually accumulates yolk, which gives the cell an opaque white appearance. The largest mature oocytes, shown here, acquire a darker cap due to accumulation of melanin pigment. The largest oocytes of X. tropicalis are about 0.8 mm in diameter, those of X. laevis about 1.4 mm, while those of the axolotl are an enormous 2.2 mm. Except for size, all three are similar in general appearance. Scale bar = 1 mm.

Figure 2: Removal of the GV from an Oocyte of X. tropicalis. Left. A small hole was made with jeweler's forceps in the darker animal pole of an oocyte and the GV was gently extruded. In this example the GV was almost free of yolk as it came out of the oocyte. Adherent yolk can be removed by sucking the GV in and out of a pipette with a tip diameter just slightly larger than the diameter of the GV itself. Right. Three GVs of X. tropicalis. These GVs were left a few minutes in a slightly acid medium (GV isolation solution adjusted to pH 5.8), which causes the nuclear envelope to swell away from the gelled nuclear contents. GVs treated in this way will not spread for cytological examination. However, such GVs are ideal for molecular analysis: the envelope can be removed with jewelers forceps, providing a sample of nuclear contents completely free of cytoplasmic contamination.17,18 Scale bar = 0.5 mm. Please click here to view a larger version of this figure.

Figure 3: GV of X. tropicalis Isolated in Oil. Left. After a small puncture was made in the oocyte near the dark animal pole, the GV began to extrude. In this case the GV came out with almost no adherent yolk. Middle. The GV is now completely free from the oocyte cytoplasm. Such GVs continue to transcribe RNA for hours. Right. After the GV is gently squashed in oil under a coverslip, nuclear organelles can be viewed by DIC, as shown here, or by phase contrast. The entire GV is much larger than the small area shown here. HLB = histone locus body with three speckles on its surface. Scale bar = 0.5 mm for first two panels, 10 µm for the third. Please click here to view a larger version of this figure.
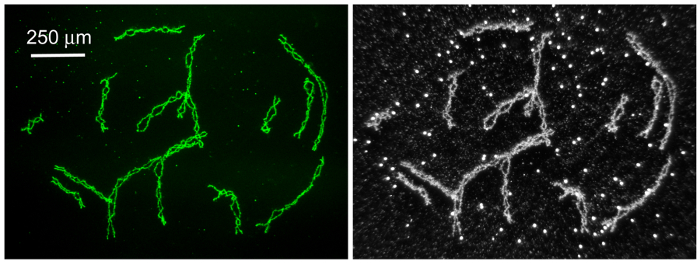
Figure 4: Lampbrush Chromosomes (LBCs) from the Axolotl A. mexicanum. Left. The 14 paired chromosomes from a single GV in prophase of the first meiotic division, immunostained with an antibody against phosphorylated RNA polymerase II. The tiny stained "dots" are histone locus bodies. Right. The same preparation viewed by darkfield illumination. One can now see the numerous unstained amplified nucleoli. Scale bar = 250 µm. Please click here to view a larger version of this figure.
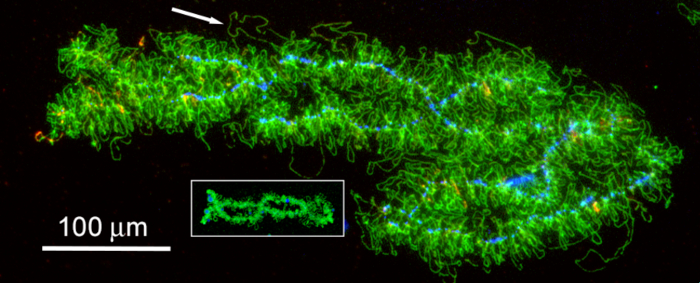
Figure 5: A Single LBC from the Axolotl A. mexicanum and the Frog X. tropicalis, at the Same Magnification. These images illustrate the extreme size difference between LBCs of a salamander and a frog (inset). The size difference correlates with the total DNA content of the genomes (about 30 Gbp for A. mexicanum vs 1.7 Gbp for X. tropicalis). Individual lateral loops (transcription units) are also much longer in the salamander than in the frog. Scale bar = 100 µm. Please click here to view a larger version of this figure.
