Magnetic cell purification of lymphocytes allows users to purify a target cell population in a relatively short amount of time. Using our depletion protocol, we were able to increase the percentage of CD8 T cells (OT-I in recombination-activating gene-1 (RAG-1)-deficient mice) from 72.8% (before purification) to 94.2% (after purification; Figure 1A)4,5. These purified lymphocytes can then be used for downstream functional assays to determine lymphocyte proliferation and signal transduction4,5. For example, we can study the in vivo T cell stimulation capacity of APCs by transferring CFSE-labeled, antigen-specific T cells into wildtype (WT) and mutant (MT) mice immunized with suitable antigen5.
In our representative experiment, we transferred purified, CFSE-labeled, ovalbumin (OVA) specific OT-I CD8 T cells into control (WT) and mutant (MT) mice and immunized these mice one day later with OVA protein. Three days after immunization, we harvested lymph nodes (proximal and distal) and spleens and analyzed the T cells by flow cytometry. CD45 allelic forms can be utilized to better separate the dividing, CFSE-labeled, donor OT-I CD8 cells from non-labeled, recipient CD8 T cells. In this example, donor and recipient cells are from CD45.1 and CD45.2 mice, respectively (Figure 1B). The CFSE levels of surviving, unstimulated OT-I T cells at this time point are used to define the peak for non-proliferating cells (Figure 1C, dotted line). Upon stimulation, the intensity of the CFSE levels in OT-I T cells will reduce by half with each division. T cell proliferation can thereby be determined by counting the number of CFSE peaks6,12. In our representative results, we do not see differences in the cross-presentation capacities of WT and MT APCs (Figure 1C, solid lines), since the T cells proliferate at similar rates in both mice.
In a separate experiment, we performed a [3H]-thymidine incorporation assay to study the cell proliferation of activated control and MT T cells. Purified WT and MT T cells were stimulated with various stimuli for 48 hr and pulsed with [3H]-thymidine for the final 8 hr. Proliferating cells in the S-phase of the cell cycle will incorporate the radiolabeled nucleotide, [3H]-thymidine, into newly synthesized deoxyribonucleic acid (DNA), therefore, using liquid scintillation counting, cell proliferation can be measured by [3H]-thymidine uptake. The number of counts per minute (c.p.m.) directly correlates with the amount of [3H]-thymidine uptake in proliferating T cells. In our representative results, the reduced number of c.p.m. obtained from MT T cells upon stimulation compared to counts from WT T cells indicates a compromised proliferative capacity of MT T cells (Figure 1D).
To determine B cell proliferative capacity, we purified splenic B cells using the described depletion strategy. Upon purification, we were able to increase the percentage of B220 B cells (WT mice) from 63.9% (before purification) to 98.4% (after purification; Figure 2A)4. Similar to purified T cells, purified splenic B cells can also be used for downstream functional assays to assess lymphocyte proliferation and signal transduction4. Subsequently, we labeled purified WT splenic B cells with CFSE and stimulated these cells in vitro using plates coated with anti-CD40 supplemented with the cytokine IL-4. Three days after stimulation, we analyzed viable, activated B cells by flow cytometry. The CFSE levels of surviving, unstimulated B cells at this time point are used to define the peak for non-proliferating cells (Figure 2B, dotted line). Similar to T cells, the intensity of the CFSE levels in B cells will reduce by half with each division6,12.
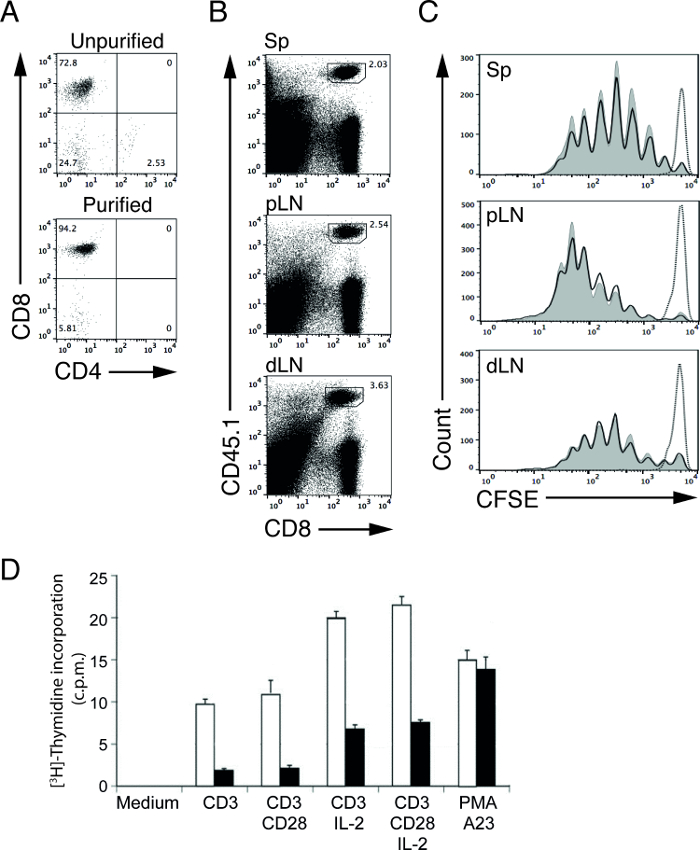
Figure 1: CFSE profiles of adoptively transferred purified OT-I T cells after immunization. (A) CD4 and CD8 staining of cells isolated from OT-I; RAG-1-deficient mice before (upper panel) and after (lower panel) non-T cell depletion. (B) Representative FACS plots of T cells from spleen (Sp), proximal lymph nodes (pLN) and distal lymph nodes (dLN). Donor OT-I CD8 T cells are CD45.1 positive. (C) Representative CFSE profiles of transferred, CFSE-labeled OT-I donor T cells from spleen (Sp), proximal lymph nodes (pLN) and distal lymph nodes (dLN) of WT (shaded curves) and MT (solid lines) recipient mice 3 days after immunization with OVA protein and LPS. Dotted line indicates OT-I T cells from WT recipient mice without immunization (PBS injected). (D) Cell proliferation of activated control or MT T cells as measured by [3H]-thymidine uptake assay. Results are presented in counts per minute (c.p.m.). Purified control (open bar) or MT (closed bar) T cells were incubated for 48 hr in medium only (medium) or in the presence of plate-bound anti-CD3 or anti-CD3 plus anti-CD28 with or without recombinant IL-2. Polyclonal cell activation was triggered by PMA (phorbol ester) and A23 (calcium ionophore). Please click here to view a larger version of this figure.
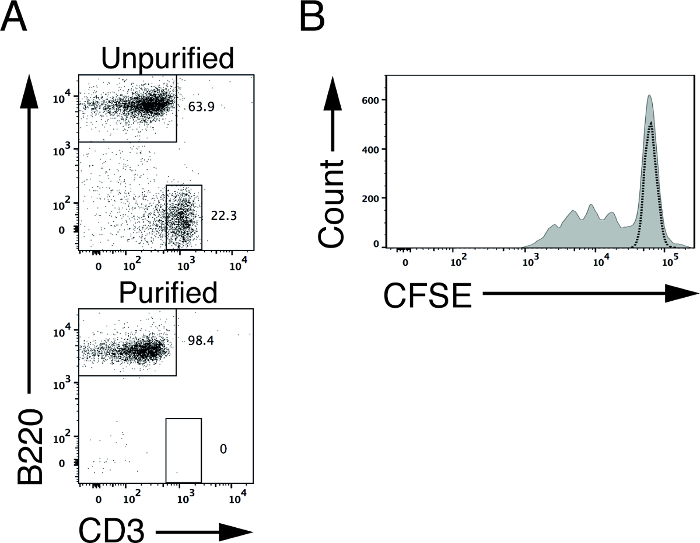
Figure 2: CFSE profile of B cells upon in vitro stimulation. (A) B220 and CD3 staining of splenic cells isolated from WT mice before (upper panel) and after (lower panel) non-B cell depletion. (B) Representative CFSE profile of CFSE-labeled WT (shaded curve) splenic B cells after 3 days of in vitro stimulation with plates coated with anti-CD40 and IL4. Dotted line indicates CFSE-labeled WT B cells without stimulation. Please click here to view a larger version of this figure.
