The photoreceptor rhodopsin, a prototypical G protein-coupled receptor (reviewed for example in reference 1), is the first phospholipid scramblase to be identified and biochemically verified 2,3. Scramblases are phospholipid transporters that increase the intrinsically slow rate of transbilayer phospholipid movement to physiologically appropriate levels in a bidirectional, ATP-independent manner 4-6. Examples of their actions can be found in the endoplasmic reticulum and bacterial cytoplasmic membrane where constitutive scrambling is needed for membrane homeostasis and growth, as well as for a variety of glycosylation pathways 5. Regulated phospholipid scrambling is needed to expose phosphatidylserine (PS) on the surface of apoptotic cells where it acts as an "eat-me"-signal for macrophages 7 and provides a procoagulant surface on activated blood platelets to catalyze the production of protein factors needed for blood clotting. In photoreceptor disc membranes, rhodopsin's scrambling activity has been suggested to counteract the phospholipid imbalance between the two membrane leaflets of the bilayer that is generated by the ATP-dependent, unidirectional lipid flippase ABCA4 4,8,910-12.
Despite the physiological importance of scramblases, their identity remained elusive until rhodopsin was reported as a scramblase in photoreceptor discs 2, members of the TMEM16 protein family were identified as Ca2+-dependent scramblases needed for PS exposure at the plasma membrane (reviewed in reference 13), and the bacterial protein FtsW was proposed as a Lipid II scramblase required for peptidoglycan synthesis 14. These discoveries were based on the reconstitution of purified proteins in liposomes and demonstration of scramblase activity in the resulting proteoliposomes using the methodology described here. Other potential scramblases 15-21 — the MurJ and AmJ proteins implicated in peptidoglycan biosynthesis, WzxE and related proteins implicated in scrambling O-antigen precursors, MprF protein needed to translocate aminoacylated phosphatidylglycerol across the bacterial cytoplasmic membrane, and Xkr8 family members that have been proposed to expose PS on the surface of apoptotic cells — remain to be tested biochemically. This highlights the importance of a robust assay to identify and characterize scramblase activity.
Here, we describe the reconstitution of purified opsin, the apoprotein of the photoreceptor rhodopsin, into large unilamellar vesicles (LUVs), and subsequent analysis of scramblase activity in the resulting proteoliposomes using a fluorescence-based assay. There are several well-described protocols available in the literature for the heterologous expression and purification of opsin, therefore we will not describe it in this protocol; we use the protocols described in Goren et al. 3 which yields FLAG-tagged, thermostable opsin at about 100 ng/µl in 0.1% (w/v) dodecylmaltoside (DDM).
Reconstitution is achieved by treating LUVs with sufficient detergent so that they swell but do not dissolve. Under these conditions, a membrane protein — supplied in the form of protein-detergent micelles — will integrate into the liposomes and become reconstituted into the liposome membrane upon detergent removal, resulting in proteoliposomes. To reconstitute opsin (obtained as a purified protein in 0.1% (w/v) DDM), LUVs are prepared from a mixture of POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]) and saturated with DDM before adding opsin and NBD-PC. The detergent is then removed by treating the sample with polystyrene beads.
The principle underlying the fluorescence-based assay is shown in Figure 1B. LUVs are symmetrically reconstituted with a trace amount of NBD-PC or other NBD-labeled fluorescent phospholipid reporter (Figure 1A). On adding dithionite, a membrane-impermeant dianion, NBD-PC molecules in the outer leaflet of the LUVs are rendered non-fluorescent as the nitro-group of NBD is reduced to a non-fluorescent amino-group. As neither NBD-PC molecules nor dithionite are able to traverse the membrane on the time-scale of the experiment (<10 min), this results in 50% reduction of the fluorescent signal. However, if the liposomes are reconstituted with a scramblase, NBD-PC molecules in the inner leaflet can scramble rapidly to the outside where they are reduced. This results in the total loss of fluorescence in the ideal case (Figure 1C).
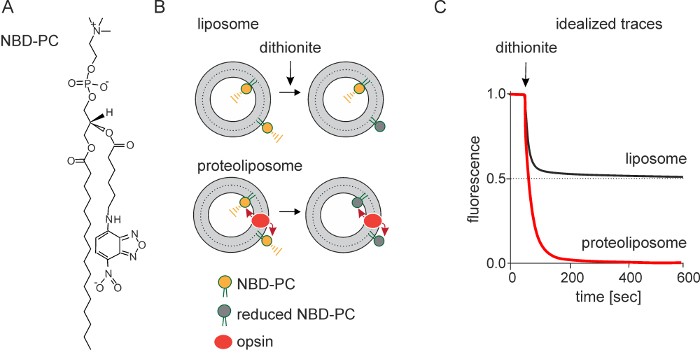
Figure 1: Schematic representation of the scramblase activity assay. The assay uses a fluorescent NBD-labeled reporter lipid; NBD-PC is shown (A). Large unilamellar vesicles are reconstituted with a trace amount of NBD-PC. Reconstitution produces symmetric vesicles, with NBD-PC distributed equally in the outer and inner leaflets. Dithionite (S2O42-) chemically reduces the nitro-group of NBD to a non-fluorescent amino-group. Treatment of protein-free liposomes with dithionite (B, top) causes a 50% reduction of fluorescence since only the NBD-PC molecules in the outer leaflet are reduced: dithionite is negatively charged and cannot cross the membrane to react with NBD-PC molecules in the inner leaflet. Dithionite treatment of opsin-containing proteoliposomes (B, bottom), i.e., scramblase-active proteoliposomes, results in 100% loss of fluorescence as opsin facilitates movement of NBD-PC between the inner and the outer leaflet. (C) shows idealized fluorescence traces obtained on treating protein-free liposomes and opsin-containing proteoliposomes with dithionite. The rate of fluorescence loss is the same in both cases indicating that the chemical reduction of NBD by dithionite is rate-limiting, and that scrambling occurs at a rate equal to or greater than the rate of the chemical reaction. Traces obtained from an actual experiment are shown in Figure 3. Please click here to view a larger version of this figure.
The methods we describe can be used to reconstitute and assay other purified proteins, as well as mixtures of membrane proteins obtained, for example, by extracting microsomes with detergent 22.
We describe the reconstitution of opsin into LUVs to characterize its scramblase activity using a fluorescence-based assay. We analyze the results to place a lower limit on the rate of opsin-mediated phospholipid scrambling and to determine the oligomeric state in which opsin functionally reconstitutes into the vesicles.
To identify optimal reconstitution conditions, it is necessary to determine empirically the amount of detergent that must be used to swell the LUVs so that they are receptive to protein insertion. Such an experiment is illustrated in Figure 2A. Aliquots of POPC:POPG LUVs are treated with different amounts of DDM for 3 hr and the absorbance of the sample is measured at 540 nm, a measure of turbidity and therefore of vesicle size. The optical density measured at 540 nm (OD540) graph shows that the vesicles swell as DDM concentration is increased from 4-6 mM, reach a saturation point at around 7 mM before starting to solubilize (corresponding to loss of turbidity and OD540 signal) as DDM is increased further. NBD-PC is added after the detergent treatment and the samples are further incubated for an hour before being treated with polystyrene beads to remove detergent. The transbilayer distribution of NBD-PC is determined by measuring fluorescence loss after adding dithionite to the vesicles (as described above). Thus, NBD-PC inserts into the outer leaflet of vesicles in the absence of detergent, indicated by its complete accessibility to dithionite. For vesicles treated with >2 mM DDM, NBD-PC is able to distribute symmetrically into both leaflets so that once DDM is removed 50% of the NBD-PC is protected from dithionite.
Figure 2B shows a similar destabilization-reconstitution experiment (redrawn from reference 2), this time tracking the reconstitution of opsin. In this experiment it can be seen that 7 mM DDM is needed for opsin reconstitution such that NBD-PC becomes quantitatively (>80%) accessible to dithionite as a result of opsin-mediated scrambling.
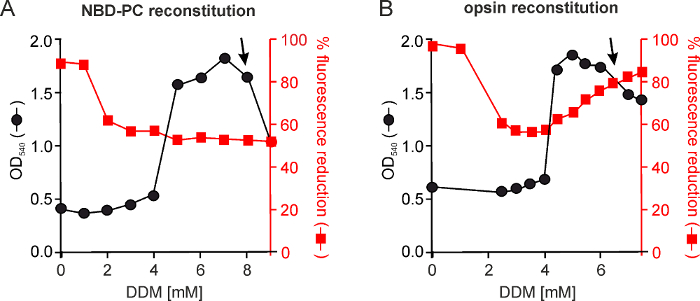
Figure 2: Reconstitution of NBD-PC and opsin into detergent-destabilized vesicles. (A) Aliquots of the vesicles are treated with a range of detergent concentrations (0-9 mM) by mixing end-over-end for 3 hr at room temperature. At the end of the incubation period the absorbance of the samples at 540 nm is measured (black line). NBD-PC (dissolved in 0.1% w/v DDM) is then added and the sample is incubated for a further 1 hr at room temperature before the detergent is removed by polystyrene beads treatment. The resulting liposomes are analyzed by the fluorescent reduction assay (red line) to determine the extent to which NBD-PC is symmetrically reconstituted. (B) As in (A) but in this experiment vesicles formed from egg phospholipids (egg PC/egg phosphatidic acid, 9:1 mol/mol) were destabilized with DDM, and opsin was added together with NBD-PC, resulting in a fluorescence reduction of about 80% once the protein is efficiently reconstituted and able to facilitate scrambling of NBD-PC (modified from reference 2). Although NBD-PC was symmetrically reconstituted at 2 mM DDM, the ideal conditions for reconstituting opsin were chosen around the peak of OD540 absorbance, i.e., the point where the vesicles were highly swollen due to intercalated detergent but not yet solubilized (indicated by the arrow). For POPC/POPG (9:1 mol/mol) vesicles, a DDM concentration of 8 mM was found to be optimal for both NBD-PC and opsin reconstitution. Please click here to view a larger version of this figure.
The extent of fluorescence reduction increases with the amount of opsin used for reconstitution. Figure 3A shows fluorescence traces obtained on adding dithionite to NBD-PC-containing protein-free liposomes (black trace) and proteoliposomes reconstituted with opsin at different protein to phospholipid ratios (PPR, in units of mg protein per mmol phospholipid, red traces); the traces have been normalized so that they all have the same initial fluorescence (Fi) value for easier visualization. Monoexponential fits of the traces indicate very similar time constants, ranging from 20-25 sec; as the rate of dithionite-mediated NBD-PC reduction in protein-free liposomes is the same as in the proteoliposomes, it is only possible to place a lower estimate on the rate of opsin-mediated scrambling (see text for more details). Figure 3B shows the analyzed data obtained from the fluorescence reduction assays to yield plots of p (≥1) scramblase (the probability of a vesicle having at least one scramblase) versus the experimentally measured PPR (related to PPR* as discussed above). Comparison of the monoexponential fit of the experimental results in hypothetical fits corresponding to opsin being reconstituted as a monomer, dimer or tetramer show that opsin reconstitutes functionally as a dimer.
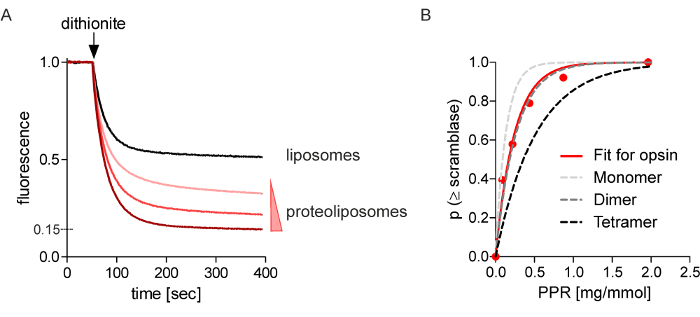
Figure 3: Scramblase activity of opsin. (A) Fluorescence traces corresponding to dithionite treatment of vesicles reconstituted with NBD-PC and opsin amounts ranging between 0-4.95 µg, indicated by the wedge. The lowest amount of opsin reconstituted corresponds to a PPR of 0.21 mg/mmol, the second highest amount to 0.43 mg/mmol and the highest amount to 1.3 mg/mmol, or 10 opsins per vesicle on average. Dithionite was added at the indicated time (arrow) and NBD fluorescence was monitored for a further 400 sec. The maximum extent of fluorescence reduction seen in this experiment was 85% whereas the average over many experiments is 82.5%. (B) Protein dependence of scrambling. Data from experiments similar to panel A were analyzed using Poisson statistics to generate a graph of the probability of vesicles having at least one scramblase (p ≥1 scramblase) versus the protein to phospholipid ratio (PPR) of the vesicles (protein was quantified post-reconstitution by Western Blot analysis 3 and phospholipid was determined by measuring inorganic phosphate released upon acid hydrolysis). The red line represents a mono-exponential fit for the data; the dashed grey lines represent mono-exponential fits for reconstitution of opsin into 170 nm-diameter vesicles as monomers, pre-formed dimers or pre-formed tetramers. As the x-axis represents the experimentally measured PPR (rather than PPR* (see main text)), the fit constants correspond to PPR rather than PPR* values, as discussed in the main text. Please click here to view a larger version of this figure.
