To determine the spatial distribution of 5hmC and 5caC in differentiating hepatic progenitors immunostained slides for these DNA modifications were imaged using a microscope.
Initial analysis of the spatial distribution of these oxi-mCs was carried out through the generation of 2.5D intensity plots (Figure 1A, 1B). The red and green peaks appeared to be well defined with the limited presence of orange peaks indicating only a small overlap of the 5caC and 5hmC signals. In agreement with these results, the profiles for 5hmC and 5caC intensities in the corresponding cell do not strongly coincide with each other (Figure 1C). This illustrates that 5caC and 5hmC exhibit distinct patterns of nuclear distribution in hepatic progenitors suggesting that Tet-dependent oxidation of 5mC leads to generation of different oxidized derivatives of this DNA modification in specific chromatin regions.
To compare the levels of 5caC and 5hmC in Daoy medulloblastoma and BXD-1425EPN ependymoma cells, immunostaining for these oxi-mCs was carried out on both samples under identical experimental conditions. Intensities of 5caC and 5hmC signals were then compared in 20 profiles generated across 3-5 cells recorded for each cell line (Figure 2A-2D). The quantification of these results revealed that BXD-1425EPN cells exhibit significantly lower levels of both 5hmC and 5caC immunostaining compared to Daoy cells (Figure 2D).
Considering the two channels red (5hmC) and green (5caC) which can be denoted as R and G respectively, the Pearson's correlation coefficient (PCC) describes the degree of spatial overlap and co-segregation of R & G assuming both marks exist in a linear relationship with each other, denoted by R statistic34. Squaring the PCC value, denoted by R2 enables the measurement of green signal intensity variation which is influenced by red signal intensity34. This is superfluous for our 5caC vs 5hmC co-localization analysis.
To examine the spatial distribution of 5caC and 5hmC in undifferentiated (Undiff) hiPSCs and hepatic endoderm 24 hours (HE24) after induction, colocalization analysis of these DNA modifications was carried out on the same confocal images, with R values for 20 nuclei analysed for each cell line (Figure 3A-3C).
The quantification of these results demonstrated that degree of colocalization is significantly higher (P < 0.05) in HE24 compared to Undiff (Figure 3c). This may be attributable to the reduced magnitude of 5caC presence and thus its reduced signal intensity in Undiff cells.
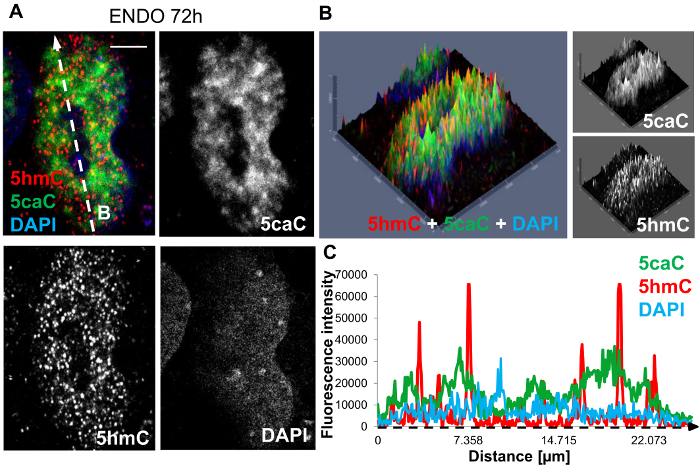
Figure 1: Immunohistochemical staining of hepatic endoderm progenitors at 72 hours post induction of differentiation. (A) Co-staining of 5hmC (red), 5caC (green) and DAPI nuclear counter stain (blue). The white dashed arrow 'B' denotes the direction of profiling sampled through the nucleus, illustrated in 1C. Scale bar represents 5 µm. Red channel: gain 800, laser 1%. Green channel: gain 750, laser 2.4%. DAPI channel: gain 750, laser 2.6%. (B) 2.5D intensity plot of 5hmC, 5caC and DAPI signals. Individual peaks represent absolute signal intensities of each pixel. Merged views and individual channels are shown. (C) The intensity profiles of 5hmC, 5caC and DAPI signals in hepatic progenitors at 72 hours post induction of differentiation. Peak intensities were recorded along the dimensions of the white dashed arrow 'B' and are indicated along the intensity profile x-axis by a black dashed arrow. Please click here to view a larger version of this figure.
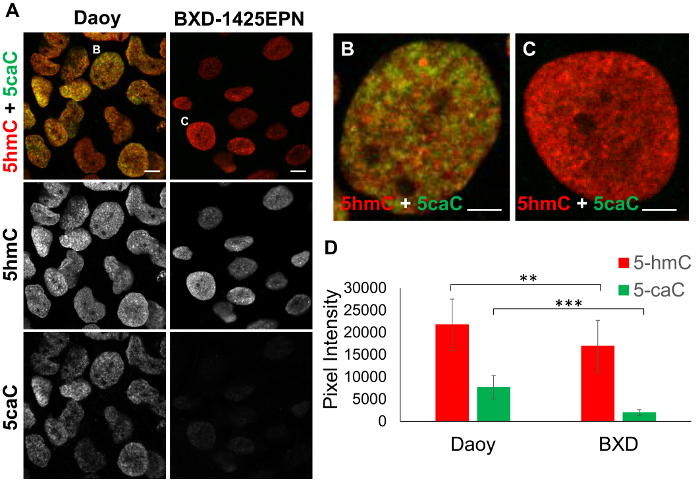
Figure 2: Analysis of 5hmC and 5caC signal intensities in Daoy medulloblastoma and BXD-1425EPN ependymoma cell lines. (A) Immunofluorescent staining of 5hmC (red) and 5caC (green) in Daoy and BXD-1425EPN cells. Images were captured with 63x oil immersion objective lens, both at the same settings. Merged views and individual channels are shown. Scale bars represent 10 µm. Red channel: gain 746, laser 1%. Green channel: gain 750, laser 2.4%. Individual (B) Daoy and (C) BXD-1425EPN cells are expanded in adjacent figures. Scale bars for (B) and (C) represent 5 µm. (D) Mean fluorescence intensity of 5hmC vs 5caC signals in Daoy and BXD-1425EPN cells (n = 20, SD). Significance was assessed using an F-test and Two-sample t-Test. Asterisks denote statistically significant p-values of ** as p <0.01 and *** as p <0.001. Please click here to view a larger version of this figure.
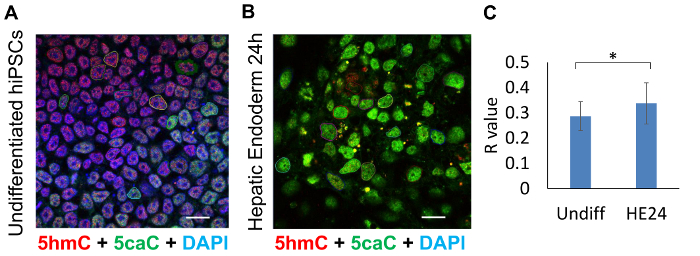
Figure 3: Colocalization analysis of 5hmC and 5caC absolute signals in undifferentiated hiPSCs (Undiff) compared to hepatic endoderm 24 hours post-differentiated progenitor cells (HE24). Confocal microscopy images of (A) undifferentiated hiPSCs and (B) HE24 cells stained for 5hmC (red), 5caC (green) and DAPI (blue). Cells were imaged with 63X oil immersion lens at identical settings. 20 encircled nuclei illustrate the cells sampled for colocalization analysis. Scale bars represent 20 µm. Red channel: gain 800, laser 1%. Green channel: gain 750, laser 2.4%. DAPI channel: gain 750, laser 2.6%. (C) Colocalization analysis of the presence of 5caC signal within range of 5hmC proximity in undiff and HE24 (n = 20, SD). Significance was assessed using an F-test and Two-sample t-Test. Asterisks denote statistically significant p-values of * p <0.05. Please click here to view a larger version of this figure.
