The structure of a 9-mer peptoid with N-terminal acetylation, Ac-Nae-(Npe-Nme)4-NH2, is shown in Figure 1. The peptoid was synthesized manually in a fritted polypropylene reaction vessel via solid phase approach. Rink amide resin (0.047 mmol, 84 mg with loading 0.56 mmol/g) is used as the solid support to yield the peptoid with an amidated C-terminus. The peptoid chain is built by multiple cycles of monomer addition. Each monomer addition cycle involves two reactions steps, bromoacetylation and displacement. The bromoacetylation is achieved by adding 0.8 M BMA solution and 0.8 M DIC solution and the reaction takes 20 min. The displacement is achieved by adding 1.0 M amine solution to the acetylation product and the reaction takes 1 h. N-terminal acetylation was carried out by adding a cocktail solution containing 92 µL of acetic anhydride, 43.5 µL of DIPEA, and 2 mL of DMF. The peptoid is cleaved off from the resin by adding a cocktail solution containing 3.8 mL of TFA, 100 µL of TIPS, and 100 µL of HPLC-grade H2O, and the reaction takes 2 h. TFA is removed in the hood by blowing in a stream of nitrogen gas until about 1 mL viscous solution is left. The peptoid product precipitates in diethyl ether and is isolated by centrifugation, and this is followed by two iterations of lyophilization. The resulting peptoid is sufficiently pure for MS/MS analysis.
The predicted fragmentation scheme for the peptoid is shown in Figure 2a, where the proton in the dashed circle indicates the "mobile proton" that would induce peptoid fragmentation during the CID experiment. The peptoid ion fragments at the amide bonds along the peptoid backbone, for which the fragmentation sites are indicated by the dashed lines. The N-terminal fragments are labeled as B-type ions and the C-terminal fragments are labeled as Y-type ions. If the fragmentation occurs at all available amide bonds, a total of eight N-terminal fragments, B1 to B8, and a total of eight C-terminal fragments, Y1 to Y8, would form. Each fragment has a corresponding m/z value that is calculated by summing up the nominal masses of all elements in the fragment. As an example, the structures and the corresponding m/z values (nominal masses) of the B4-ion and the Y5-ion are shown in Figure 2b. The chemical formula for the B4 ion is C31H42N5O6+, and the nominal mass is calculated by the equation (31 x 12) + (42 x 1) + (5 x 14) + (6 x 16) = 580. Since B4 is a singly charged ion, the m/z value would be 580/1 = 580. In Figure 2b, the structure of the B4-ion is a simplified form (see discussion section for details). The calculated m/z values (nominal masses) of all fragment ions from B1-B8 and from Y1-Y8 are given in Table 1.
The mass analysis includes two processes. The first process is to perform the full scan mass spectrometry analysis of the peptoid sample. This result indicates whether the sample contains a measurable amount of the peptoid, and the relative purity of the sample. The full scan mass spectrum of the peptoid ion is shown in Figure 3, where the m/z values are rounded to the nearest whole number. The peak at m/z 1,265 corresponds to the protonated peptoid, and the peak at m/z 1,287 corresponds to the sodium ion adduct of the peptoid. The two peaks at m/z 633 and m/z 644 correspond to doubly protonated and mixed protonated-sodiated peptoids, respectively. These results suggest that the peptoid sample is sufficiently pure for carrying out MS/MS analysis.
The second process in mass analysis is to perform the MS/MS experiment on the protonated peptoid at m/z 1,265. This process includes isolating the precursor peptoid ion by the first quadrupole unit, fragmenting the peptoid ion in CID, and sorting the fragment ions according to their m/z values by the third quadrupole unit. The resulting spectrum is shown in Figure 4, where the m/z values are rounded to the nearest whole number. The peak at m/z 1,265 corresponds to the protonated peptoid. The other peaks at lower m/z values correspond to the fragment ions from the peptoid ion. The fragment ions are assigned as either the B-ion or the Y-ion by comparing their m/z values with those predicted (shown in Table 1) based on the peptoid fragmentation scheme (shown in Figure 2). Seven Y-ions (Y2 to Y8) and seven B-ions (B1 to B7) are formed with observable abundances. Notice that the abundance of the Y-ions is much higher than that of most B-ions.
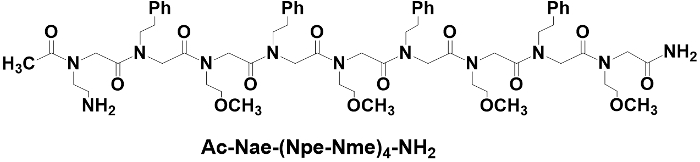
Figure 1: Chemical structure of the peptoid Ac-Nae-(Npe-Nme)4-NH2. The peptoid was synthesized manually via the solid phase approach. Please click here to view a larger version of this figure.
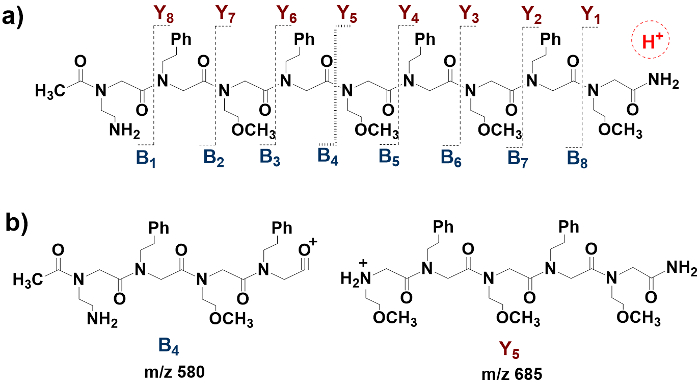
Figure 2: Fragmentation scheme for the protonated peptoid Ac-Nae-(Npe-Nme)4-NH2. The peptoid ion fragments at the amide bonds along the peptoid backbone to produce a series of N-terminal fragments called B-ions and a series of C-terminal fragments called Y-ions. a) Predicted fragmentation scheme for the protonated peptoid Ac-Nae-(Npe-Nme)4-NH2. The dashed lines indicate the fragmentation sites, the symbols B1 to B8 indicate the N-terminal fragments, and the symbols Y1 to Y8 indicate the C-terminal fragments; b) Sample fragmentation with schematic structures. The structures show the B4-ion and the Y5-ion with corresponding m/z values, respectively. The m/z values are calculated by summing up the nominal masses of the elements in the structures. Please click here to view a larger version of this figure.
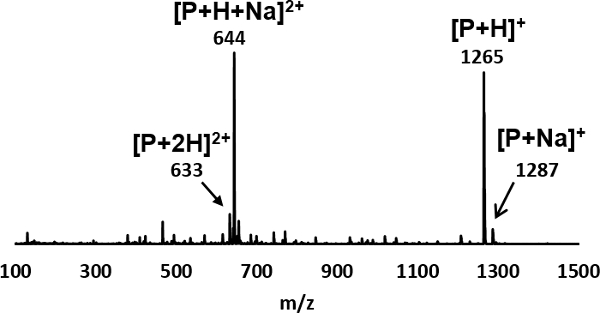
Figure 3: Full scan mass spectrum of the peptoid Ac-Nae-(Npe-Nme)4-NH2, where the m/z values are rounded to the nearest whole number. The peak at m/z 1,265 corresponds to the peptoid ion, [P+H]+, and the peak at m/z 1,287 corresponds to sodium ion adduct of the peptoid, [P+Na]+. The two peaks at m/z 633 and m/z 644 correspond to the doubly charged peptoid, [P+2H]2+ and [P+H+Na]2+, respectively. Please click here to view a larger version of this figure.
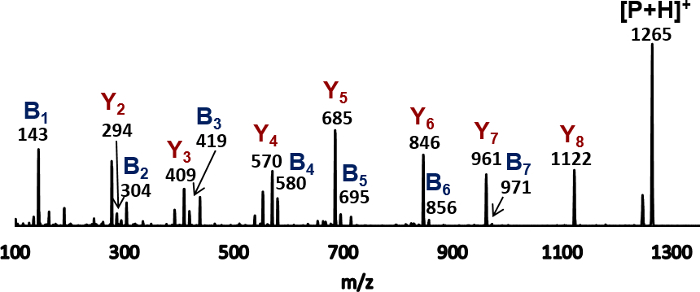
Figure 4: MS/MS spectrum of the protonated peptoid Ac-Nae-(Npe-Nme)4-NH2 labeled with assigned fragments. The peak at m/z 1,265 corresponds to the protonated peptoid, [P+H]+, and the peaks with lower m/z values correspond to the fragment ions. The B- and Y-ions are assigned by comparing their m/z values with those calculated based on the fragmentation scheme of the peptoid ion. Please click here to view a larger version of this figure.
| B-ion | m/z Value (Nominal Mass) | Y-ion | m/z Value (Nominal Mass) |
| B1 | 143 | Y1 | 133 |
| B2 | 304 | Y2 | 294 |
| B3 | 419 | Y3 | 409 |
| B4 | 580 | Y4 | 570 |
| B5 | 695 | Y5 | 685 |
| B6 | 856 | Y6 | 846 |
| B7 | 971 | Y7 | 961 |
| B8 | 1132 | Y8 | 1122 |
Table 1: Theoretical m/z values calculated based on the predicted fragmentation scheme of the protonated peptoid Ac-Nae-(Npe-Nme)4-NH2. The B-ion and the Y-ion indicate the corresponding fragment ions of the N-terminus and the C-terminus, respectively. Each m/z value (nominal mass) is calculated by summing up the nominal masses of the elements in that fragment ion.
