Using the technique of selective pressure incorporation, Trp-66 in the chromophore triad of ECFP (and Trp-57, the only other Trp residue in ECFP) can be replaced by 4-amino-Trp, thereby generating the red-shifted GdFP with distinct spectral properties. Mass spectrometry must be used to demonstrate the desired stoichiometric integration of the non-canonical amino acid into the protein, with results shown in Figure 1. Afterwards, we provide data from microscopy, UV-Vis absorption spectroscopy as well as steady-state and time- and wavelength-resolved fluorescence spectroscopy to characterize the properties of the GdFP fluorophore with a focus on the pH dependence of the spectra.
To confirm the exchange of the two Trp residues in ECFP by 4-amino-Trp, mass spectrometric analysis is carried out. Figure 1 shows a representative deconvoluted ESI-MS spectrum of GdFP. While wild-type ECFP has a calculated protein mass of 28,283.9 Da after the chromophore maturation, the corresponding mass of GdFP is 28,313.9 Da. The deconvoluted ESI-MS spectrum of GdFP exhibits a main mass peak at 28,314.1 ± 0.1 Da, which deviates from the theoretical value by less than 10 ppm. Being within the typical accuracy range for this type of analysis25, this confirms the incorporation of the ncAA via SPI (experimental value for wild-type ECFP: 28,283.7 Da).
Figure 2 shows confocal fluorescence imaging microscopy (CFIM) images of bacterial cells expressing ECFP, EGFP, EYFP and GdFP upon resuspension of bacteria in PBS buffer. All images were acquired on a microscope equipped with a UV objective and laser excitation at about the same energy for each sample.
Figure 3A shows an overlay of CFIM images of E. coli bacteria expressing various FPs including GdFP, always monitored with very similar excitation energy (wavelengths as in Figure 2). Figure 3B shows the chromophore structures of the FP variants shown. Regarding the brightness of GdFP compared to ECFP (fluorescence quantum yield φ = 0.4), EGFP (φ = 0.6) and EYFP (φ = 0.6) it is important to note that for GdFP, a broader acquisition range of the fluorescence light (30 nm) was used in contrast to 20 nm used for all other species, in order to adjust the intensity of the images to similar values. With a slightly lower extinction coefficient and a reduced quantum yield as a consequence of unique photophysical properties, the brightness of GdFP is lower compared to the other FPs shown.
The absorption spectrum of ECFP (Figure 3C) has two characteristic maxima at 434 nm and 452 nm. In contrast, GdFP is characterized by one broad red-shifted absorption band with the maximum at 466 nm. The absorption of EGFP is further red-shifted to 488 nm. However, due to the much larger Stokes shift of GdFP (108 nm) compared to ECFP (41 nm) and EGFP (20 nm), the emission spectrum of GdFP is the most red-shifted of all three GFP derivatives investigated here (Figure 3D). While the fluorescence emission of ECFP shows two characteristic maxima at 475 nm and 505 nm, EGFP has one broad main emission band peaking at 508 nm (λmax) with a slight shoulder at 540 nm. The fluorescence of GdFP appears at about 565 nm (λmax.) (Figure 3D). Its emission spectrum contains a small contribution of wild-type ECFP which is also visible as a small shoulder at 475 nm. This small ECFP fraction is synthesized before induction during the SPI procedure, as described33.
Figure 3E shows the pH-dependent changes in the absorption spectrum of GdFP. For a pH change from 8 to 5, the emission maximum shifts slightly to the red and a slight broadening of the absorption band is observed. However, the reduction of the absorption amplitude is only about 10 % between pH 8 and pH 5, indicating that the ground state properties of the GdFP chromophore are very weakly modified by pH.
The time resolved fluorescence emission monitored by single photon counting is shown in Figure 4. The decay curves monitored in the spectral channels centered at 550 nm and 600 nm (Figure 4A) exhibit a slightly faster fluorescence decay at 600 nm compared to the decay at 550 nm. The results of a global fit of the fluorescence decay curves with two exponential components results in two spectrally distinguishable fluorescence decay components with time constants of 1.0 ns and 3.3 ns (Figure 4C and D).
The fluorescence emission of GdFP strongly depends on pH, as it is typical for many fluorescent protein variants of the GFP family. Figure 4B compares the fluorescence emission of GdFP between pH 5 and pH 8, which clearly shows a decrease in the fluorescence intensity at lower pH, while the spectral characteristics stay constant.
The decay-associated spectra (DAS)39 of GdFP (Figure 4C and D) are characterized by two distinct emission bands. The contribution of the slow 3.3 ns component is more pronounced in the short wavelength range around 550 nm (60 %) with minor contribution of the faster component (40 %). At 600 nm, both components have about the same amplitude. Upon a shift from pH 7 (Figure 4C) to pH 6 (Figure 4D), the spectral characteristics of the DAS hardly change and the time constants from the global fitting routine are also the same (the accuracy of the DAS time constants is about ± 0.15 ns). However, the difference in the absolute amplitudes of the two DAS components is clearly apparent, which fully accounts for the reduced fluorescence emission amplitude upon the same pH shift in Figure 4B.
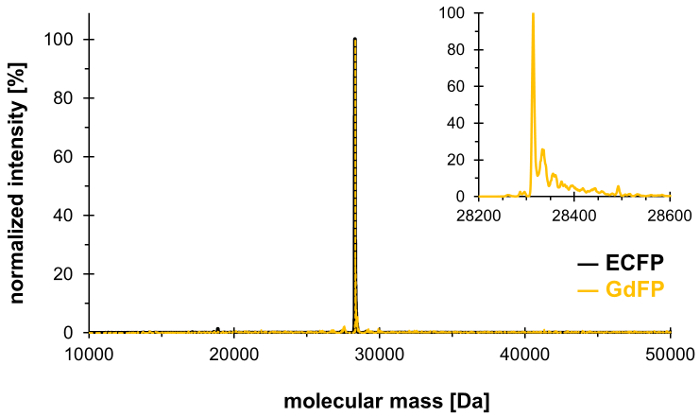
Figure 1: Representative deconvoluted ESI-MS spectrum of GdFP. The ESI-MS spectrum of GdFP (gold color, magnified plot shown as inset) shows a main peak at 28314.1 Da (calculated value 28313.9 Da). The spectrum for wild-type ECFP is shown in black. Please click here to view a larger version of this figure.
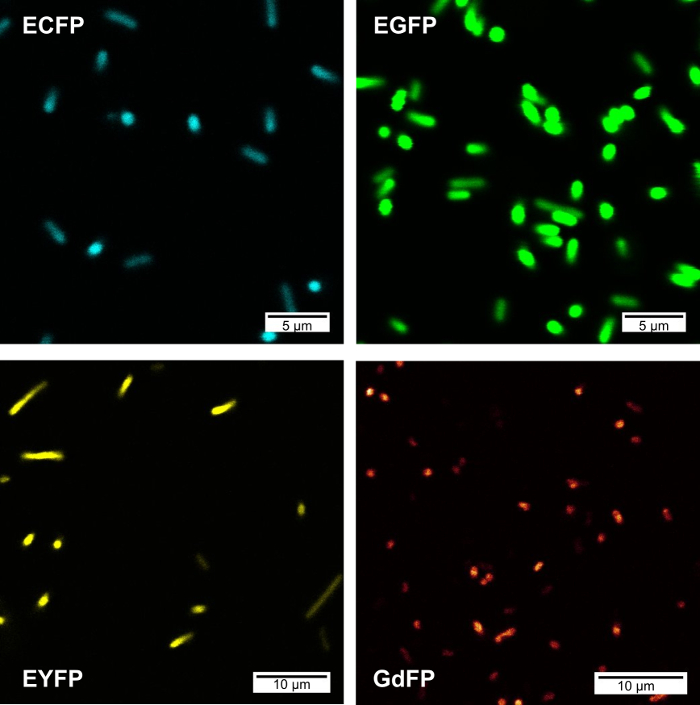
Figure 2: Confocal fluorescence microscopy images from bacterial populations expressing various FPs. The following wavelength settings were used for image acquisition: ECFP (λex = 457 nm, detection: 461-480 nm), EGFP (λex = 488 nm, detection: 495-515 nm), GDFP (λex = 476 nm, detection: 560-590 nm), EYFP (λex = 514 nm, detection: 520-530 nm). Please click here to view a larger version of this figure.
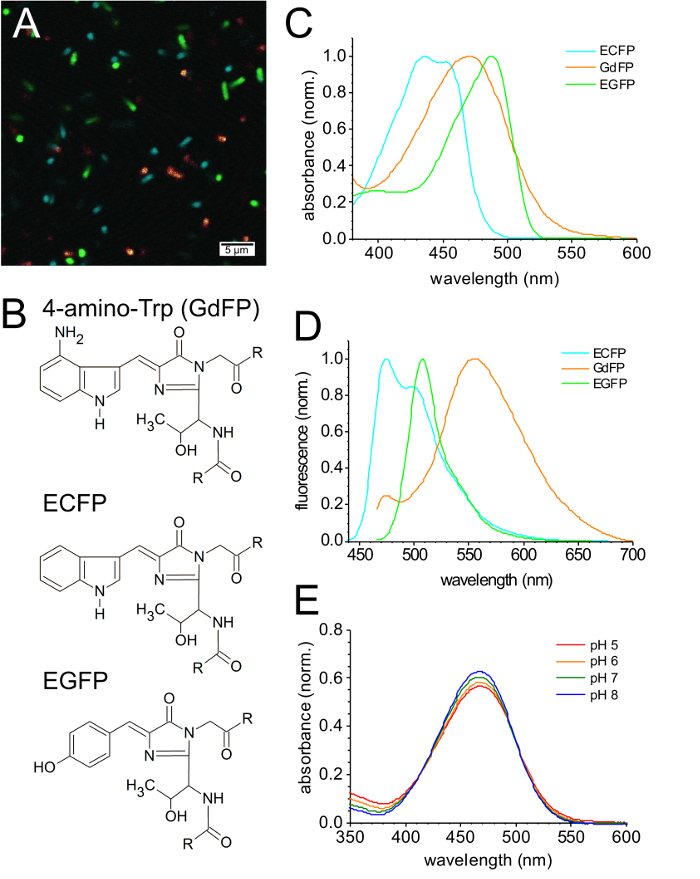
Figure 3: Spectral properties of GdFP. (A) CFIM image of a mixture of bacterial cells expressing ECFP, EGFP and GdFP after the resuspension of bacteria in PBS buffer. (B) Chromophore structures of GdFP (with 4-amino-Trp in place of residue 66), the parental ECFP (with Trp at position 66) and EFGP (with Tyr at position 66). (C) Comparison of the normalized absorption spectra of GdFP, ECFP and EGFP, whereas in (D), the normalized fluorescence emission spectrum of ECFP (excitation at 430 nm) is compared to the fluorescence emission spectra of EGFP and GdFP (both excited at 450 nm). (E) pH-dependence of the absorption spectra (normalized absorption at 280 nm). Please click here to view a larger version of this figure.
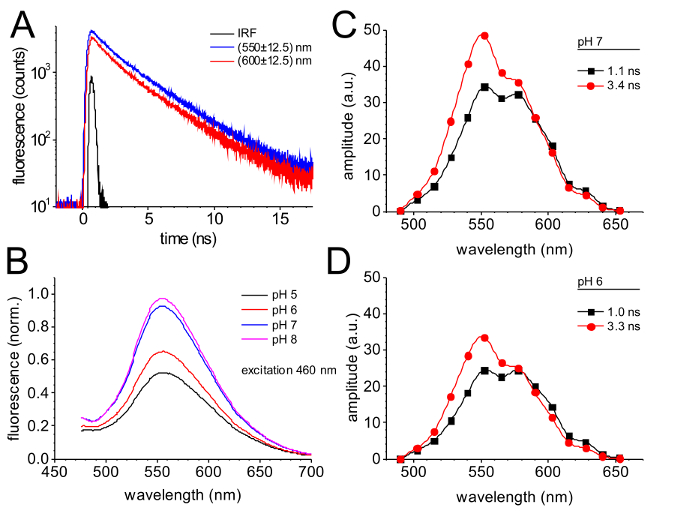
Figure 4: Time-resolved fluorescence of GdFP. (A) Fluorescence decay of GdFP monitored by time- and wavelength-resolved single photon counting in the spectral channels centered at 550 nm and 600 nm (± 12.5 nm) after excitation with 470 nm laser pulses. The instrumental response function (IRF) provides information about the time resolution of the used setup. (B) Variation of the emission spectrum of GdFP dependent on pH (excitation at 460 nm). (C, D) Decay-associated spectra (DAS) of GdFP at pH 7 (C) and pH 6 (D) determined after deconvolution of time- and wavelength-resolved fluorescence decays and global fitting of the decays in all channels by a global set of two exponential functions with linked time constants. Please click here to view a larger version of this figure.
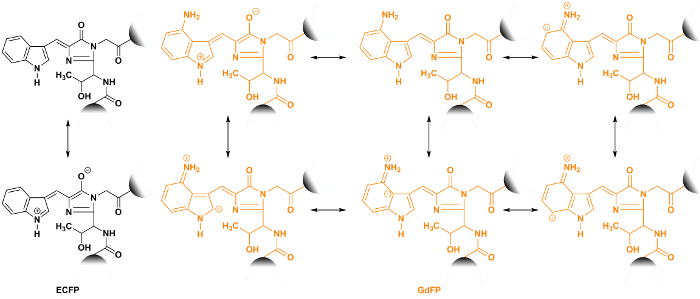
Figure 5: Structures of the intramolecular charge transfer of ECFP (black) and GdFP (gold) chromophores. The increase in size of the chromophore system by the good electron donor of an amino group as part of the ncAA enables the formation of more mesomeric structures to achieve resonance stabilization of the excited state. The connection points to the FP scaffold are shown as semicircles. Please click here to view a larger version of this figure.
| Stock solution | concentration, solvent | Note | |
| 20% D-glucose | 200 g/L D-glucose in ddH2O | sterilize by filtration through a 0.45 µm pore size syringe filter | |
| indole | 50 mM in isopropanol | ||
| 4-amino-indole | 50 mM in 20 % ethanol (20 mL ethanol in a final volume of 100 mL filled up with ddH2O) | ||
| IPTG | 1 M in ddH2O | ||
| L-tryptophan | 15 mM dissolved in ddH2O using 1 M HCl (add HCl dropwise under stirring until powder is dissoved) | ||
| lysozyme | 50 mg/mL in ddH2O | ||
| DNase I | 1 mg/mL in ddH2O | ||
| RNase A | 1 mg/mL in ddH2O | ||
| Amp100 | 100 mg/mL ampicillin in ddH2O | ||
| sodium-dodecylsulfate (SDS) | 200 g/L in ddH2O | ||
| ammonium sulfate ((NH4)2SO4) | 1 M in ddH2O | sterilize by autoclaving | |
| potassium dihydrogen phosphate (KH2PO4) | 1 M in ddH2O | sterilize by autoclaving | |
| di-potassium hydrogen phosphate (K2HPO4) | 1 M in ddH2O | sterilize by autoclaving | |
| magnesium sulfate (MgSO4) | 1 M in ddH2O | sterilize by autoclaving | |
| D-glucose | 1 M in ddH2O | sterilize by filtration through a 0.45 µm pore size syringe filter | |
| sodium chloride (NaCl) | 5 M in ddH2O | sterilize by autoclaving | |
| calcium chloride (CaCl2) | 1 g/L | sterilize by filtration through a 0.45 µm pore size syringe filter | |
| iron(II) chloride (FeCl2) | 1 g/L | sterilize by filtration through a 0.45 µm pore size syringe filter | |
| thiamine | 10 g/L | sterilize by filtration through a 0.45 µm pore size syringe filter | |
| biotin | 10 g/L | sterilize by filtration through a 0.45 µm pore size syringe filter | |
| trace elements mix | copper sulfate (CuSO4), zinc chloride (ZnCl2), manganese chloride (MnCl2), ammonium molybdate ((NH4)2MoO4); each 1 mg/L in ddH2O | sterilize by filtration through a 0.45 µm pore size syringe filter | |
| 19 amino acids mix | 1.) Dissolve 0.5 g L-phenylalanine and 0.5 g L-tyrosine in 100 ml ddH2O with dropwise addition of 1 M HCl under stirring until powder is dissolved. | ||
| 2.) Weigh out 0.5 g of each of the remaining L-amino acids (except L-tryptophan). Mix with 22 mL fo 1 M KH2PO4 and 48 mL of 1 M K2HPO4. Add ddH2O to about 800 mL. Stir until the solution becomes clear. | |||
| 3.) Add the dissolved L-phenylalanine and L-tyrosine from step 1.) and adjust the volume to 1 L with ddH2O. | |||
| 4.) Sterilize the amino acid mixture by vacuum filtration with a bottle top filter unit. | |||
| Buffers and Media | Composition/Preparation | ||
| SDS loading dye buffer, 5x concentrated | 0.25 M Tris pH 6.8, 50 % v/v glycerol, 0.25 % w/v bromphenol blue, 0.5 M didhiothreitol (DTT; alternatively 5 % β-mercaptoethanol), 10 % w/v sodium-dodecylsulfate (SDS) | ||
| binding buffer | 50 mM sodium dihydrogenphosphate (NaH2PO4), 500 mM NaCl, 10 mM imidazole, pH 8 | ||
| elution buffer | 50 mM sodium dihydrogenphosphate (NaH2PO4), 500 mM NaCl, 250 mM imidazole, pH 8 | ||
| dialysis buffer | 50 mM sodium dihydrogenphosphate (NaH2PO4), 150 mM NaCl, 100 mL/L glycerol, pH 8 | ||
| MS buffer | 10 mM Tris-HCl, pH 8 | ||
| new minimal medium containing 19 L-amino acids except L-tryptophan (NMM19) | Mix all stock solutions to obtain the following final concentrations: 7.5 mM (NH4)2SO4, 1.7 mM NaCl, 22 mM KH2PO4, 50 mM K2HPO4, 1 mM MgSO4, 20 mM D-glucose, 50 mg/L of 19 amino acids mix, 1 µg/L CaCl2, 1 µg/L FeCl2, 10 µg/L thiamine, 10 mg/L biotin, 0.01 mg/L trace elements mix | ||
| LB medium | Composition: 10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl, pH 7.0 in ddH2O | ||
| Preparation: | |||
| 1.) Weigh out 50 g tryptone, 25 g yeast extract, 5 g NaCl into a 1 L glass bottle. | |||
| 2.) Add ddH2O up to ~800 mL and dissolve components under stirring. | |||
| 3.) Measure pH and adjust to pH 7 by dropwise addition of 1 M HCl or 1 M NaOH, if necessary. Add ddH2O up to 1 L. | |||
| 4.) Sterilize by autoclaving, check for volume loss afterwards and add sterile ddH2O to compensate if necessary. Store at 4 °C until use. | |||
| LB agar plates | Composition: 10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl, 15 g/L agar-agar, pH 7.0 in ddH2O | ||
| Preparation: | |||
| 1.) Weigh out 50 g tryptone, 25 g yeast extract, 5 g NaCl, 7.5 g agar-agar into a 1 L glass bottle. | |||
| 2.) Add ddH2O up to 500 mL and dissolve components under stirring. | |||
| 3.) Measure pH and adjust to pH 7 by dropwise addition of 1 M HCl or 1 M NaOH, if necessary. Add ddH2O up to 1 L. | |||
| 4.) Sterilize by autoclaving, check for volume loss afterwards and add sterile ddH2O to compensate, if necessary. (Note: LB agar can be stored at 4 °C until use for preparation of LB agar plates. Carefully melt solidified agar using a microwave) | |||
| 5.) When the solution is still warm (30-40 °C), add ampicillin to a final concentration of 100 µg/mL | |||
| 6.) Pour about 15 mL of the liquid from step 5.) into a sterile 10 cm Petri dish under sterile conditions. When the agar is solidified, plates can be stored for 1 week at 4 °C until use. | |||
| phosphate-buffered saline (PBS) | Composition: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 1 mM CaCl2, 0.5 mM MgCl2, pH 7. Sterilize by autoclaving or filtration. | ||
Table 1: Stock solution and buffer.
